Аномальный кариотип – как он влияет на фертильность?
Воспользуйтесь знаниями наших специалистов
- Главная страница
- Знания
Возвращение к списку статей
15 novembris 2021 | 1,5 минуты чтения
Трудности в зачатии и протекании беременности могут быть вызванными генетическими изменениями (мутациями). Хотя генетические изменения кажутся незаметными, они могут вызывать множество нарушений, а также, могут неосознанно передаваться планируемому потомству. Поэтому стоит сделать генетическую диагностику. Одним из основных методов является определение кариотипа.
Что такое кариотип?
Кариотип представляет собой набор хромосом, в которых хранится весь генетический материал человека. Нормальным должно быть 22 пары аутосомных хромосом (идентичных как мужским, так и женским), отвечающим за наследственные гены, и 1 пара половых хромосом.
Показания к обследованию кариотипа
Хотя причин и симптомов генетических изменений может быть много, стоит обратить особое внимание на некоторые из них. Показания к исследованию кариотипа включают:
- аномальное анатомическое строение половых органов;
- возникновение генетических заболеваний в семье,
- дисфункция репродуктивной системы (гипогонадизм)
- повторяющиеся выкидыш или гибель плода,
- потомство с хромосомными дефектами,
- отсутствие явной причины бесплодия, не выявленной при других методах диагностики;
- азооспермия или олигоспермия,
- нарушения менструального цикла или первичная аменорея,
- преждевременная менопауза.
У Вас есть вопросы? Свяжитесь с нами!
Ход изучения и интерпретация результатов
Тестирование кариотипа простое и не требует предварительной подготовки – оно предполагает взятие образца периферической крови (обычно из вены), который затем анализируется на количество хромосом в клетках и их структуру.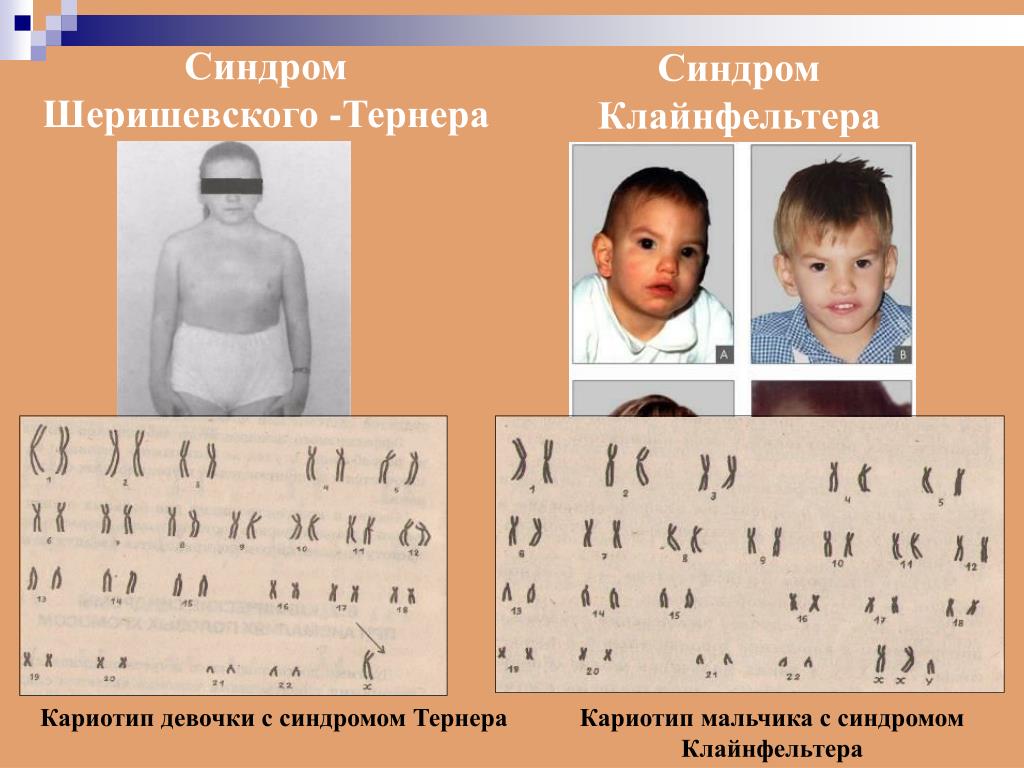
Результат теста считается правильным, если он показывает:
- мужчины: 46 аутосомных хромосом и одна пара XY,
- женщины: 46 аутосомных хромосом и одна XX пара.
Различают следующие изменения кариотипа:
- дефекты, связанные с количеством хромосом (аутосомомная хромосомная трисомия, такая как синдром Дауна, синдром Эдвардса или гендерная хромосомная трисомия, такая как синдром синдром Клайнфельтера или синдром Джейкобса),
- дефекты, связанные со структурой хромосом (транслокации – движение хромосом, инверсии – изменение части хромосомы, делеции – исчезновение хромосомных фрагментов, дубликация– дублирование фрагментов хромосом.
Неправильный кариотип – что дальше?
Неправильный кариотип не означает невозможность наступления здоровой беременности и родить потомство без генетических дефектов. В этом случае требуется генетическая консультация – что позволит оценить потенциальные шансы пары на рождение здорового ребенка, а также разработать соответствующий план лечения.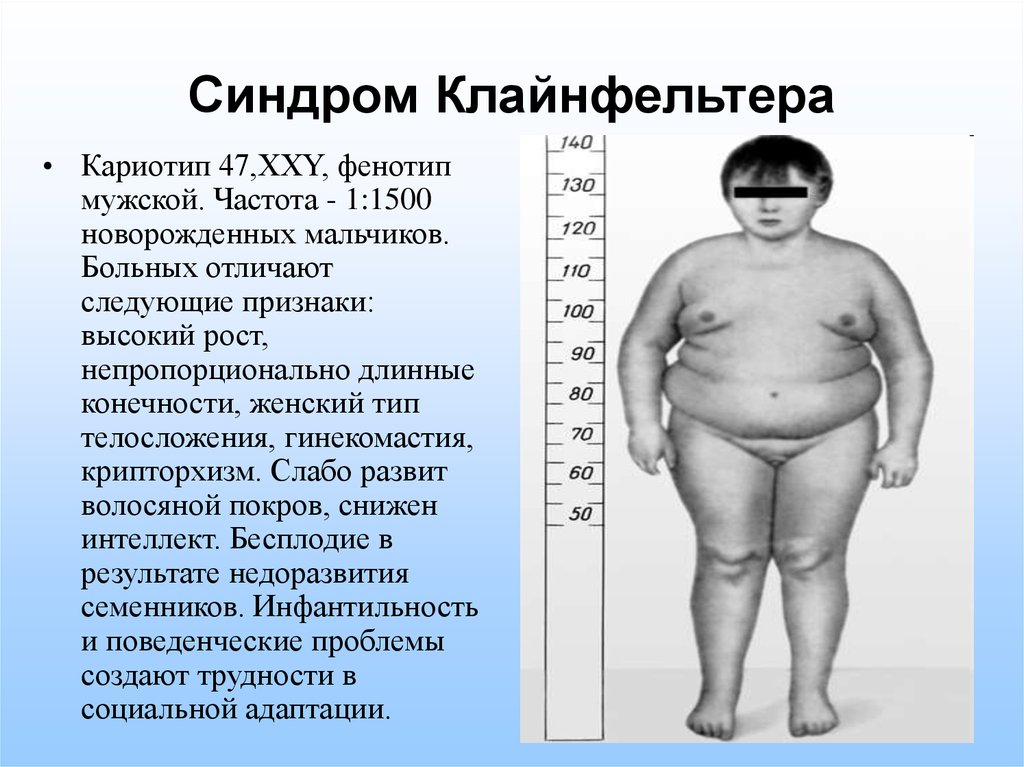 Кроме того, иногда рекомендуется проводить предимплантационную диагностику ПГС (PGT-A) или ПГД (PGT-M) в случае медицинского оплодотворения. Это позволит определить, имеют ли полученные эмбрионы генетические дефекты.
Кроме того, иногда рекомендуется проводить предимплантационную диагностику ПГС (PGT-A) или ПГД (PGT-M) в случае медицинского оплодотворения. Это позволит определить, имеют ли полученные эмбрионы генетические дефекты.
Посмотрите также
- Клиника лечения бесплодия
- Запишитесь на прием
- ЭКО
- Как происходит внутриматочная инсеминация (ВМИ)
- Анализ спермы
Хромосомы — Reproduktionsmedizin München im Tal
Хромосомные изменения
В ядре каждой отдельной клетки тела содержится вся генетическая информация, «упакованная» в хромосомы. В каждой клетке нашего тела находятся от 50 000 до 100 000 генов, которые распределяются по 46 хромосомам. Эти 46 хромосом образуют 23 пары. Они отличаются по размеру и структуре. Последними идут хромосомы X и Y. Они определяют пол индивидуума и поэтому также называются половыми хромосомами, или гоносомами.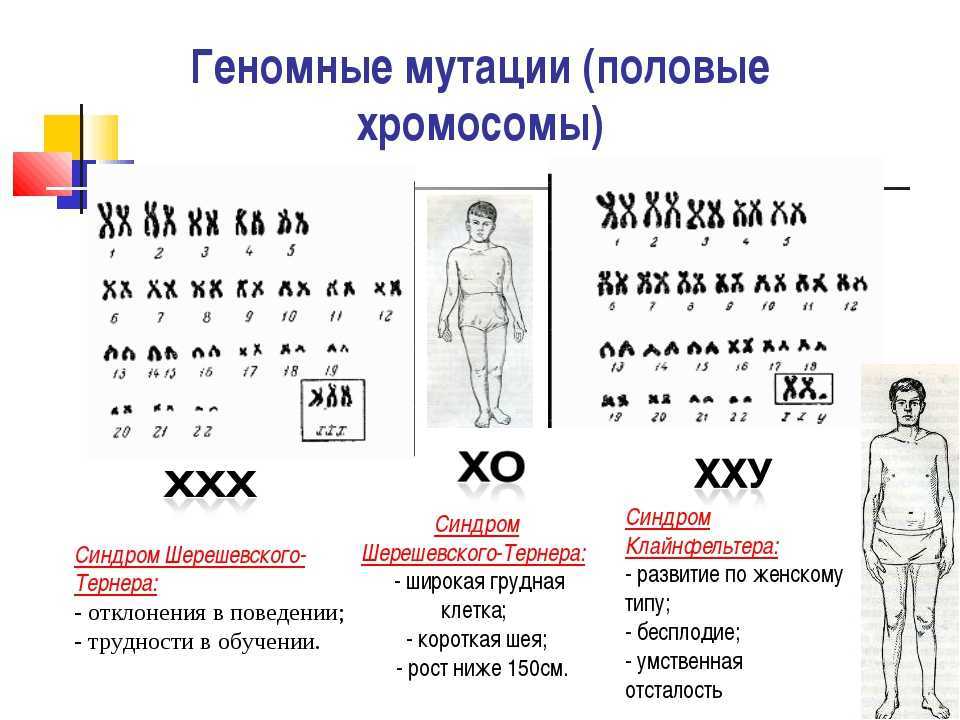
У женщин в ядрах клеток две хромосомы Х, u мужчин – одна хромосома Х и одна хромосома Y. Кариотип (от греческого karyon – ядро и typos – тип, форма) – это совокупность признаков полного набора хромосом; кариотип представляет собой количество и структуру хромосом человека.
Пример:
46, XX – нормальный женский кариотип;
46, XY – нормальный мужской кариотип
Из этого определения видно, что имеется 46 хромосом – и что речь идёт об индивидуумах мужского или женского пола.
Если количество или структура хромосом изменяются, это может привести к потере или увeличению генетического материала, т.е. так называемой несбалансированной хромосомной перестройке. Если генетический материал не изменяется в количественном плане, говорят о сбалансированной хромосомной перестройке.
Сбалансированная хромосомная перестройка
Если структура и/или количество хромосом изменяются без потери или увеличения генетического материала, то говорят о сбалансированной хромосомной перестройке. Такая перестройка не влияет на клетки организма и, следовательно, не имеeт клинической значимости. Однако в случае созревания зародышевых клеток (развитие зрелых яйцеклеток или сперматозоидов) может произойти возникновение несбалансированных яйцеклеток или сперматозоидов (зародышевые, или половые, клетки, гаметы).
Такая перестройка не влияет на клетки организма и, следовательно, не имеeт клинической значимости. Однако в случае созревания зародышевых клеток (развитие зрелых яйцеклеток или сперматозоидов) может произойти возникновение несбалансированных яйцеклеток или сперматозоидов (зародышевые, или половые, клетки, гаметы).
В этом случае возникшая в результате оплодотворения первая клетка развивающегося организма (зигота) несёт в себе несбалансированный набор хромосом, что вызывает патологию в развитии эмбриона. Это приводит либо к отсутствию беременности, потому что развивающийся эмбрион не может укрепиться в слизистой матки, либо к выкидышам или рождению детей с пороками развития и умственной отсталостью.
У мужчин сбалансированная хромосомная перестройка изменения хромосом также может привести к азоо- или олигозооспермии и, следовательно, к бесплодию
Несбалансированная хромосомная перестройка
Человек, клетки которого несут несбалансированный вариант кариотипа, в большинстве случаев развивается в соответствии с собственной генетической программой, что находит отражение в определенных особенностях внешности, аномалиях в развитии органов и умственной отсталости.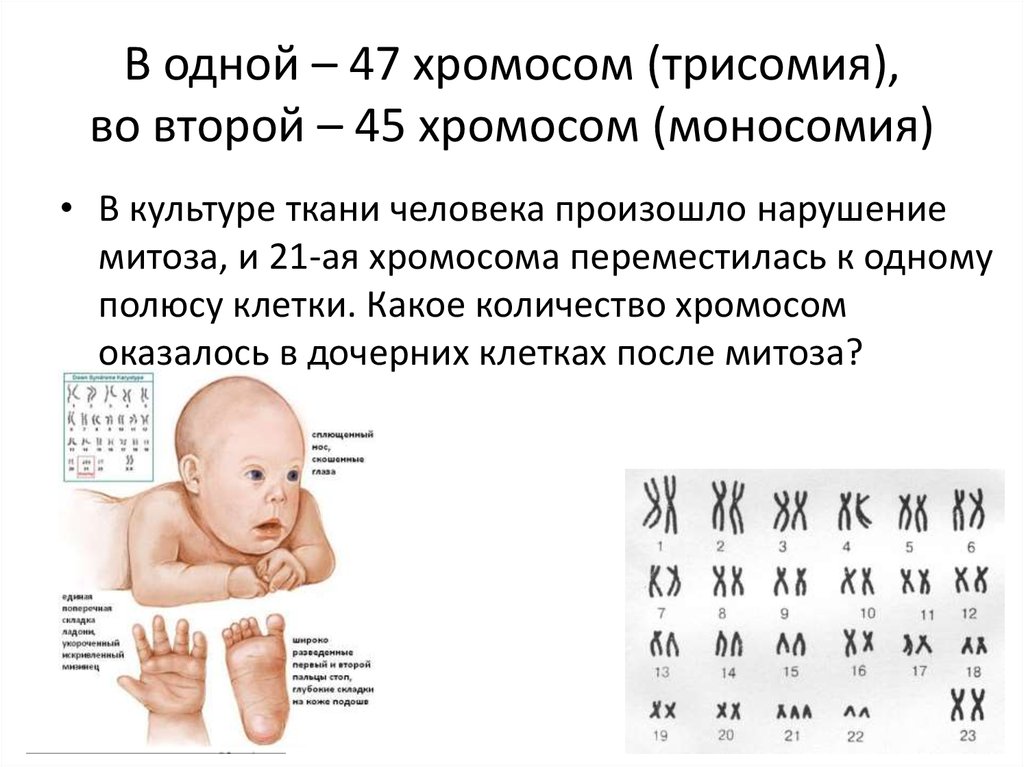
Если затронуто количество половых хромосом, то речь идёт о несбалансированном наборе хромосом, которые приводят к определённым неярко выраженным изменениям внешности. Носители несбалансированных перестроек (хромосомных мутаций, или аберраций), как правило, фенотипически нормальны. Эта особенность структуры их хромосом можно обнаружить только проведя хромосомный анализ, например, из-за нереализованного желания иметь детей.
К наиболее частым несбалансированным хромосомным перестройкам относятся:
кариотип 47, XXY (синдром Клайнфельтера)
Мальчики, у которых в кариотипе присутствует дополнительная Х-хромосома, обычно не отличаются патологией после рождения и во взрослом возрасте ведут нормальную жизнь. Из-за атрофии яичек они не способны производить потомство. В некоторых случаях у мужчин с кариотипом 47, XXY за счёт репродуктивной медицины могут быть дети.
Кариотип 45, Х (синдром Тёрнера)
Женщины с кариотипом 45, X отличаются маленьким ростом и некоторыми особенностями внешности.
 Иногда наблюдаются некоторые патологии органов, такие как сердечная недостаточность или дисфункция щитовидной железы. Ещё в детском возрасте вместо яичников у них развиваются тяжи соединительной ткани. Такие женщины стерильны.
Иногда наблюдаются некоторые патологии органов, такие как сердечная недостаточность или дисфункция щитовидной железы. Ещё в детском возрасте вместо яичников у них развиваются тяжи соединительной ткани. Такие женщины стерильны.У некоторых только несколько клеточных ядер характеризуются набором хромосом с одной недостающей Х-хромосомой. Остальные ядра с нормальным набором хромосом. Фертильность таких женщин в той или иной мере ограничена.
Кариотип 47, XXX
Женщины с этим набором хромосом здоровы и фертильны, но могут передать своим детям по наследству лишнюю Х-хромосому. В этом случае в кариотипе их сыновей набор хромосом 47, XXY (см. выше), а у дочерей тот же набор хромосом, что и у матери.
 Иногда наблюдаются некоторые патологии органов, такие как сердечная недостаточность или дисфункция щитовидной железы. Ещё в детском возрасте вместо яичников у них развиваются тяжи соединительной ткани. Такие женщины стерильны.
Иногда наблюдаются некоторые патологии органов, такие как сердечная недостаточность или дисфункция щитовидной железы. Ещё в детском возрасте вместо яичников у них развиваются тяжи соединительной ткани. Такие женщины стерильны.Мужское бесплодие | Генетические аномалии или мужское бесплодие
Развитие экстракорпорального оплодотворения (ЭКО) позволило многим парам завести семьи, которые в противном случае они не смогли бы создать самостоятельно. В то же время эта технология позволила исследователям изучить генетический состав самых ранних стадий эмбрионов. Эти достижения дают представление о связи между генетикой и бесплодием и о том, как дефекты (мутации) в определенных генах могут привести к мужскому или женскому бесплодию. Вполне возможно, что многие случаи необъяснимого бесплодия однажды обнаружат, что они имеют четкую генетическую основу.
Эти достижения дают представление о связи между генетикой и бесплодием и о том, как дефекты (мутации) в определенных генах могут привести к мужскому или женскому бесплодию. Вполне возможно, что многие случаи необъяснимого бесплодия однажды обнаружат, что они имеют четкую генетическую основу.
За последние два десятилетия вспомогательной репродукции стало известно, что некоторые случаи тяжелого мужского бесплодия явно связаны с генными делециями, мутациями или хромосомными аномалиями.
Хромосомная аномалия
У некоторых мужчин с очень тяжелым мужским фактором бесплодия при анализе хромосом крови (известном как «кариотип») обнаруживается дополнительная Х-хромосома. То есть вместо кариотипа 46 XY у них кариотип 47 XXY. Это состояние известно как «синдром Клайнфельтера» и может привести к неспособности достичь половой зрелости или даже при достижении половой зрелости у этих мужчин часто бывает мужское бесплодие. Некоторые мужчины с синдромом Клайнфельтера могут забеременеть с помощью экстракорпорального оплодотворения (ЭКО) с внутрицитоплазматической инъекцией сперматозоидов (ИКСИ). До сих пор мы не наблюдаем повышенного риска синдрома Клайнфельтера или других хромосомных аномалий у потомства, полученного в этих случаях.
До сих пор мы не наблюдаем повышенного риска синдрома Клайнфельтера или других хромосомных аномалий у потомства, полученного в этих случаях.
Микроделеции Y-хромосомы
В последние годы также было обнаружено, что у некоторых мужчин с очень серьезным низким количеством сперматозоидов обнаруживаются делеции в определенной части их Y-хромосомы, известной как ген DAZ. Их кариотип нормальный (46 XY), но внимательное изучение Y-хромосомы показывает, что некоторые участки хромосомы отсутствуют. У части этих мужчин не будет восстанавливаемой спермы в эякуляте или при операции на яичках, и донорская сперма является единственным вариантом. При других делециях гена DAZ присутствует небольшое количество сперматозоидов и возможно зачатие с помощью ЭКО-ИКСИ. В этих случаях потомки мужского пола, которые всегда будут наследовать Y-хромосому своего отца, также будут иметь эту делецию и сами будут бесплодными.
Мутации одного гена
Мутация одного гена в гене кистозного фиброза (CF) связана с отсутствием части трубы («семяпровода»), которая ведет от яичка к уретре в половом члене. Эти мужчины обычно являются носителями мутации гена муковисцидоза и сами не болеют кистозным фиброзом. Сперму можно извлечь из яичек у этих мужчин для использования в ЭКО с ИКСИ, но крайне важно, чтобы их жена (или поставщик яйцеклеток) также была полностью протестирована на мутации муковисцидоза, иначе существует значительный риск рождения ребенка с муковисцидозом. .
Эти мужчины обычно являются носителями мутации гена муковисцидоза и сами не болеют кистозным фиброзом. Сперму можно извлечь из яичек у этих мужчин для использования в ЭКО с ИКСИ, но крайне важно, чтобы их жена (или поставщик яйцеклеток) также была полностью протестирована на мутации муковисцидоза, иначе существует значительный риск рождения ребенка с муковисцидозом. .
Для мужчин с общим числом подвижных спермиев менее 5 миллионов сперматозоидов рекомендуется тестирование на генетические состояния, чтобы эти мужчины или пары могли быть осведомлены о генетических проблемах и о том, как эти проблемы могут повлиять на их потомство.
Мужское бесплодие, связанное с аберрантным кариотипом, 47,XYY: четыре клинических случая | Журнал дел
- Отчет о делах
- Открытый доступ
- Опубликовано:
- Фаеза Эль-Датори 1 и
- Хани М Эльшейха 2
11 тыс. обращений
26 цитирований
2 Альтметрика
Сведения о показателях
Abstract
История вопроса
47, XYY-синдром представляет собой половую хромосомную аномалию, наблюдаемую у людей, с распространенностью 0,1% рождений мужского пола. Аномалии половых хромосом чаще связаны с мужским бесплодием.
Аномалии половых хромосом чаще связаны с мужским бесплодием.
История болезни
Мы представляем здесь четыре случая бесплодных мужчин с азооспермией или тяжелой олигозооспермией, посещающих клинику генетики и репродукции. Хромосомный анализ лимфоцитов периферической крови показал конституциональный кариотип 47, XYY. С помощью флуоресцентной гибридизации in situ (FISH) было подтверждено наличие дополнительной Y-хромосомы, что подтверждает цитогенетические данные.
Заключение
Синдром 47,XYY встречается относительно редко и может быть пропущен клинически из-за его различных клинических проявлений. Точная диагностика этого конституционального кариотипа оказывает ценную помощь в консультировании и раннем лечении пациентов, которым проводится оценка фертильности.
История вопроса
Синдром XYY представляет собой анеуплоидию половых хромосом, при которой мужчина получает дополнительную Y-хромосому, образуя кариотип 47,XYY. Эта хромосомная аномалия встречается у одного из 1000 живорождений мужского пола в общей популяции, но чаще у бесплодных популяций [1]. В большинстве случаев фенотипические особенности остаются нормальными, что затрудняет дальнейшее лечение, основанное на специфических требованиях, связанных с определенной этиологией. Тем не менее, мальчики в возрасте 47 лет XYY имеют повышенный риск проблем с поведением, повышенную скорость роста в раннем детстве, высокий рост, легкую неспособность к обучению и задержку речевых и языковых навыков [2]. Здесь мы сообщаем о четырех случаях, показывающих синдром XYY, из 132 исследованных бесплодных мужчин.
Эта хромосомная аномалия встречается у одного из 1000 живорождений мужского пола в общей популяции, но чаще у бесплодных популяций [1]. В большинстве случаев фенотипические особенности остаются нормальными, что затрудняет дальнейшее лечение, основанное на специфических требованиях, связанных с определенной этиологией. Тем не менее, мальчики в возрасте 47 лет XYY имеют повышенный риск проблем с поведением, повышенную скорость роста в раннем детстве, высокий рост, легкую неспособность к обучению и задержку речевых и языковых навыков [2]. Здесь мы сообщаем о четырех случаях, показывающих синдром XYY, из 132 исследованных бесплодных мужчин.
Методы
Пациенты
Четыре случая были отобраны из 132 мужчин с бесплодием, посещавших клинику генетики и фертильности при академической больнице медицинского факультета Университета Мансура, Египет, и клинический генетик определил, что они могут иметь хромосомную аномалию. Пациенты были опрошены об их историях и их репродуктивных проблемах, семейном происхождении и возможном кровном родстве. За интервью последовал медицинский осмотр с целью выявления анатомических проблем. Уретральную жидкость и сперму проверяли на микробные инфекции. Гормональный профиль с количественным анализом ФГС, ЛГ и тестостерона проводился у всех больных.
За интервью последовал медицинский осмотр с целью выявления анатомических проблем. Уретральную жидкость и сперму проверяли на микробные инфекции. Гормональный профиль с количественным анализом ФГС, ЛГ и тестостерона проводился у всех больных.
Обычный цитогенетический анализ
Образцы крови собирали у всех пациентов в пробирки с гепарином. Образцы периферической крови больного культивировали в течение 72 часов в среде RPMI-1640 с добавлением эмбриональной бычьей сыворотки и фитогемагглютинина. Цитогенетический анализ проводили всем пациентам с использованием методики GTG banding [3]. Было проанализировано не менее 30 метафаз. Лучшие метафазы фотографировали для определения кариотипа больных. Кариотипы описаны по ISCN 9.5 номенклатура [4].
FISH
Мы провели FISH с использованием X- и Y-центромерных зондов с флуоресцентной меткой в соответствии с рекомендациями производителя. Анализ проводили с использованием эпифлуоресцентного микроскопа Zeiss Axioplan, оснащенного соответствующими фильтрами.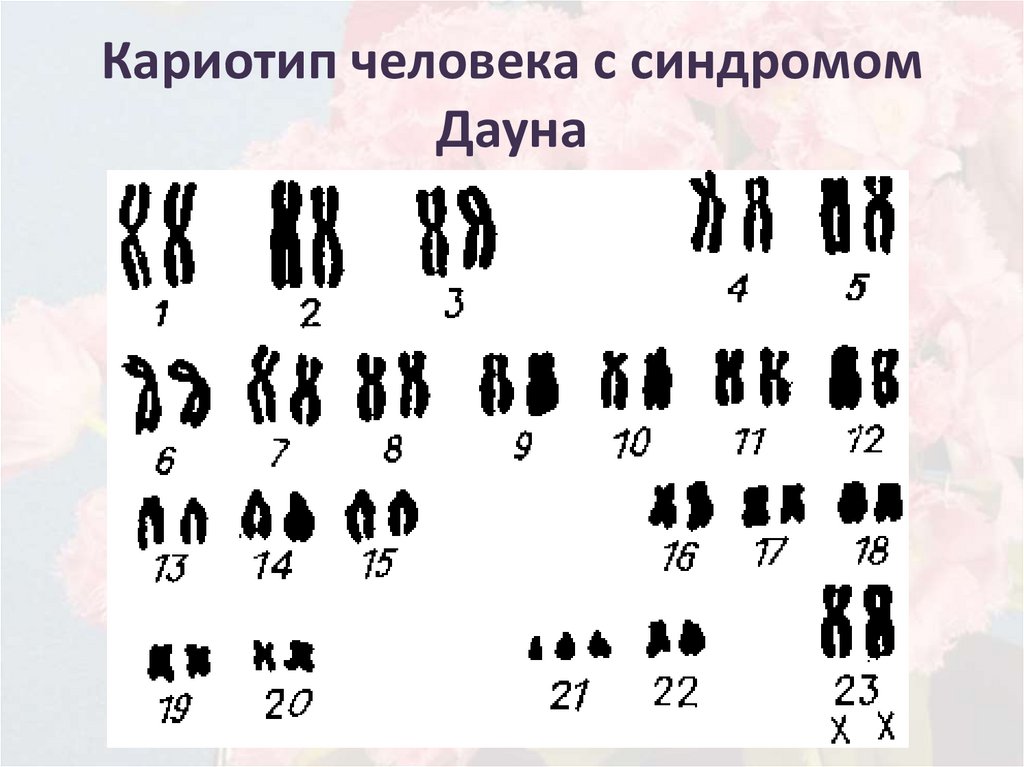 Изображения были получены с использованием программного обеспечения для анализа изображений Cytovision V2.81 (Applied Imaging International, Сан-Хосе, Калифорния, США).
Изображения были получены с использованием программного обеспечения для анализа изображений Cytovision V2.81 (Applied Imaging International, Сан-Хосе, Калифорния, США).
Результаты
Описание случая
Случай 1
31-летний мужчина поступил в больницу по поводу бесплодия. Пациент был старшим из трех нормальных братьев и сестер, рожденных от кровнородственных здоровых родителей. У него были нормальные фенотипические черты с ничем не примечательной историей болезни. Он был высокого роста (рост 193 см; вес 74 кг) при размахе рук 206 см. Эндокринологические исследования показали повышение уровней ФСГ и ЛГ (26,5 и 16,9 мМЕ/мл соответственно) и очень низкий уровень тестостерона (1,8 нг/мл). Анализ спермы выявил азооспермию.
Случай 2
В поликлинику обратился мужчина 30 лет с жалобами на бесплодие в течение 4 лет. В семейном анамнезе членов с аналогичными заболеваниями не было. Родители были двоюродными братьями. Физикальное обследование показало нормального мужчину ростом 187 см и весом 71 кг. Объем обоих яичек составил 14 мл. Измерение уровня гормонов в сыворотке показало нормальные значения ЛГ (4,2 мМЕ/мл) и тестостерона (6,8 нг/мл) с повышенным уровнем ФСГ (21,3 мМЕ/мл). В анализах семенной жидкости выявлена выраженная олигозооспермия с концентрацией сперматозоидов 0,7×10 9 .0034 6 /мл (4,9 × 10 6 /эякулят), подвижность 24% и нормальная морфология 5%.
Объем обоих яичек составил 14 мл. Измерение уровня гормонов в сыворотке показало нормальные значения ЛГ (4,2 мМЕ/мл) и тестостерона (6,8 нг/мл) с повышенным уровнем ФСГ (21,3 мМЕ/мл). В анализах семенной жидкости выявлена выраженная олигозооспермия с концентрацией сперматозоидов 0,7×10 9 .0034 6 /мл (4,9 × 10 6 /эякулят), подвижность 24% и нормальная морфология 5%.
Случай 3
30-летний мужчина с 4-летним первичным бесплодием вследствие олигозооспермии 1,3 × 10 6 /мл (8,5 × 10 6 /эякулят), 18% нормальная подвижность и 4% нормальная морфология . Он первый мальчик, рожденный от некровных родителей с двумя нормальными братьями. Нет семейной истории аналогично пораженных членов. Результаты анализа крови включали высокий уровень ФСГ и ЛГ (19,3 и 17,3 мМЕ/мл соответственно) и низкий уровень тестостерона (2,8 нг/мл).
Случай 4
40-летний мужчина обратился с жалобами на первичное бесплодие в течение 3 лет. Он самый младший мальчик, рожденный от некровного брака с двумя нормальными братьями. Не было никаких соответствующих выводов относительно его прошлой истории и привычек. История семьи ничем не примечательна. Результаты физического осмотра и ультразвукового исследования его мочеполовой системы были нормальными, с нормальным объемом яичек. Результаты по гематологическим и биохимическим показателям были нормальными. Уровни ФСГ и ЛГ были высокими (14,5 и 18,6 мМЕ/мл соответственно), а уровень тестостерона находился в пределах нижней границы нормы (3,6 нг/мл). Два анализа спермы продемонстрировали олигозооспермию с количеством сперматозоидов 2,5 × 10 9 .0034 6 /мл (8 × 10 6 /эякулят), 25% нормальная подвижность и 4% нормальная морфология.
Не было никаких соответствующих выводов относительно его прошлой истории и привычек. История семьи ничем не примечательна. Результаты физического осмотра и ультразвукового исследования его мочеполовой системы были нормальными, с нормальным объемом яичек. Результаты по гематологическим и биохимическим показателям были нормальными. Уровни ФСГ и ЛГ были высокими (14,5 и 18,6 мМЕ/мл соответственно), а уровень тестостерона находился в пределах нижней границы нормы (3,6 нг/мл). Два анализа спермы продемонстрировали олигозооспермию с количеством сперматозоидов 2,5 × 10 9 .0034 6 /мл (8 × 10 6 /эякулят), 25% нормальная подвижность и 4% нормальная морфология.
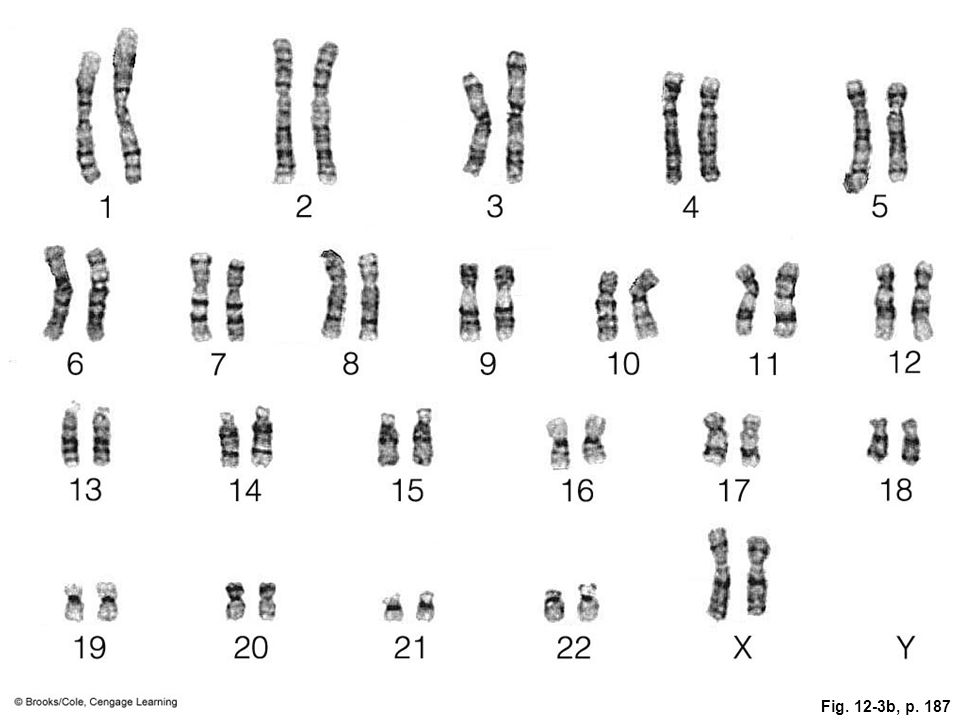
Цитогенетические особенности
Хромосомные исследования с традиционным цитогенетическим анализом выявили числовые хромосомные аномалии у четырех пациентов. GTG-бэндинг выявил аномальный немозаичный кариотип 47,XYY в метафазных клетках у всех четырех пациентов (рис. 1). Наличие дополнительной Y-хромосомы было подтверждено анализом FISH с использованием меченых флуоресценцией X- и Y-центромерных зондов (рис. 2 и 3).
2 и 3).
Кариограмма мужчин с синдромом 47,XYY .
Изображение в полный размер
Рисунок 2Метафазный анализ FISH показывает два зеленых сигнала центромера Y-хромосомы и один красный сигнал центромера Х-хромосомы, что подтверждает кариотип XYY .
Изображение в полный размер
Рисунок 3Интерфазный анализ FISH, показывающий два зеленых сигнала Y-хромосомы и один красный сигнал X-хромосомы, подтверждающий кариотип XYY .
Изображение в полный размер
Обсуждение и заключение
Было высказано предположение о наличии генетического компонента человеческого бесплодия, хотя ни специфические аномалии, ни их генетический механизм передачи полностью не определены. Целью настоящего отчета было представить четыре случая пациентов мужского пола, поступивших в отделение генетики и фертильности университетской больницы Мансура из-за проблем с бесплодием.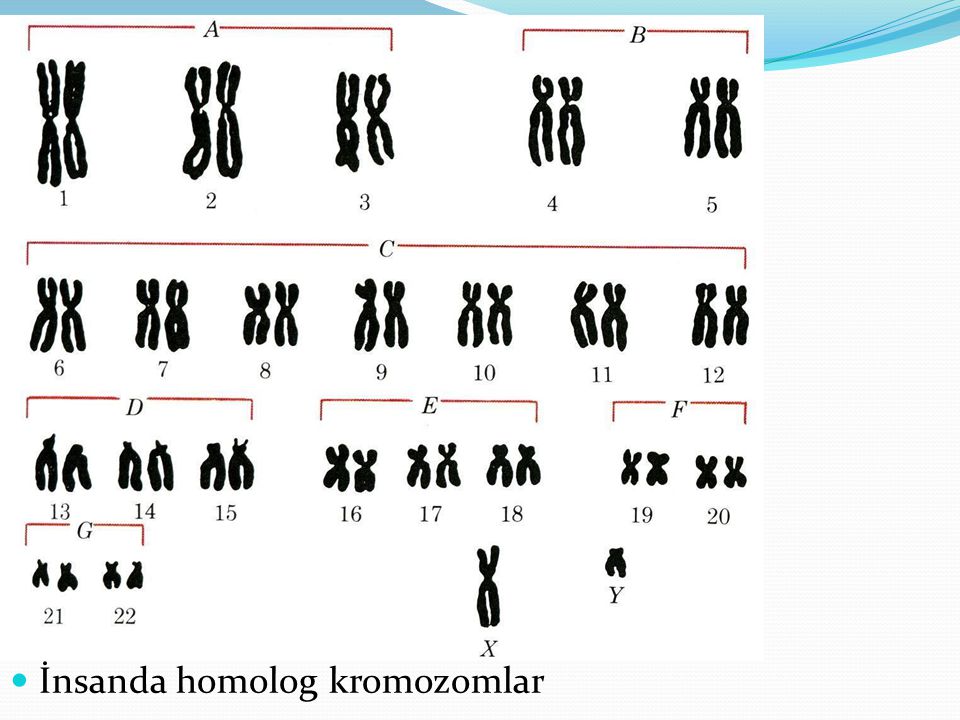 Цитогенетический анализ с использованием метода GTG-бэндинга выявил у всех больных кариотип 47,XYY. Хотя цитогенетические анализы иногда утомительны, они очень важны для выявления различных синдромов. Кроме того, успех постановки клинического диагноза с помощью цитогенетических методов можно повысить с помощью FISH и других дополнительных методов молекулярной биологии. Последнее в сочетании с обычным кариотипированием может в значительной степени преодолеть ограничения обычного бэндинга в точной диагностике и интерпретации малозаметных или сложных хромосомных аномалий. При обоих подходах в нашем исследовании был поставлен окончательный диагноз хромосомного расстройства 47, XYY. Информация, полученная с помощью таких методов, служит основой для принятия решения о необходимом клиническом ведении и генетическом консультировании пациентов, нуждающихся в этой услуге.
Цитогенетический анализ с использованием метода GTG-бэндинга выявил у всех больных кариотип 47,XYY. Хотя цитогенетические анализы иногда утомительны, они очень важны для выявления различных синдромов. Кроме того, успех постановки клинического диагноза с помощью цитогенетических методов можно повысить с помощью FISH и других дополнительных методов молекулярной биологии. Последнее в сочетании с обычным кариотипированием может в значительной степени преодолеть ограничения обычного бэндинга в точной диагностике и интерпретации малозаметных или сложных хромосомных аномалий. При обоих подходах в нашем исследовании был поставлен окончательный диагноз хромосомного расстройства 47, XYY. Информация, полученная с помощью таких методов, служит основой для принятия решения о необходимом клиническом ведении и генетическом консультировании пациентов, нуждающихся в этой услуге.
Анамнез всех пациентов ничем не примечательный. В семейном анамнезе не было ни врожденных пороков развития, ни воздействия наркотиков или токсичных агентов окружающей среды.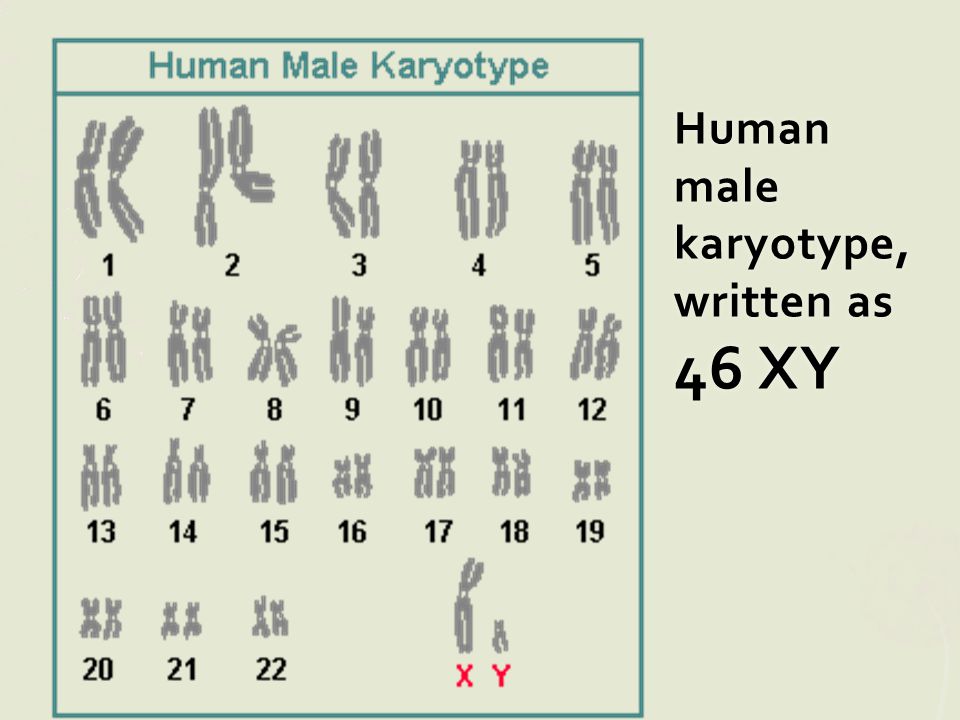 Обычные анализы не выявили инфекционной причины их бесплодия. Уровень тестостерона в норме у мужчин 47, XYY [5], но у пациентов в нашем исследовании был низкий или нормальный уровень тестостерона и высокий или нормальный уровень ФСГ и ЛГ. Анализ спермы показал аномальный профиль. Аномалии спермы могут быть вызваны несколькими причинами, такими как микроделеции Y-хромосомы и мутации гена CFTR [6, 7], но ни у одного из четырех пациентов не было обнаружено ни одной из них.
Обычные анализы не выявили инфекционной причины их бесплодия. Уровень тестостерона в норме у мужчин 47, XYY [5], но у пациентов в нашем исследовании был низкий или нормальный уровень тестостерона и высокий или нормальный уровень ФСГ и ЛГ. Анализ спермы показал аномальный профиль. Аномалии спермы могут быть вызваны несколькими причинами, такими как микроделеции Y-хромосомы и мутации гена CFTR [6, 7], но ни у одного из четырех пациентов не было обнаружено ни одной из них.
Было высказано предположение, что мужская субфертильность человека может иметь семейный компонент [8]. Из представленных данных следует, что бесплодие может иметь генетическую основу, поскольку все четыре пациентки происходили из семей с кровнородственными браками, но оценка взаимосвязи между кровным родством и этой хромосомной аномалией или возможность того, что кровное родство может влиять на фенотипические характеристики. Как правило, 47,XYY не наследуется, но обычно возникает как случайная ошибка разделения хромосом при формировании сперматозоидов, приводящая к образованию сперматозоидов с дополнительной копией Y-хромосомы. Если одна из этих своеобразных сперматозоидов вносит вклад в генетическую структуру ребенка, у ребенка будет дополнительная Y-хромосома в каждой из клеток организма [9].]. В некоторых случаях добавление дополнительной Y-хромосомы происходит в результате нерасхождения во время клеточного деления во время постзиготического митоза в раннем эмбриональном развитии. Это может создать мозаику 46,XY/47,XYY [9].
Если одна из этих своеобразных сперматозоидов вносит вклад в генетическую структуру ребенка, у ребенка будет дополнительная Y-хромосома в каждой из клеток организма [9].]. В некоторых случаях добавление дополнительной Y-хромосомы происходит в результате нерасхождения во время клеточного деления во время постзиготического митоза в раннем эмбриональном развитии. Это может создать мозаику 46,XY/47,XYY [9].
На протяжении десятилетий известно, что цитогенетические аномалии являются важной причиной мужского бесплодия, а хромосомные аномалии чаще наблюдаются у мужчин с азоо- и/или олигозооспермией, чем в общей популяции. Мужчины с кариотипом 47, XYY, как правило, фертильны, и нет доказательств передачи дополнительной Y-хромосомы их потомству, поскольку лишняя Y-хромосома удаляется во время мейоза; и некоторые зародышевые клетки XYY могут завершать мейоз и продуцировать зрелые анеуплоидные сперматозоиды [10]. Однако чаще они встречаются у бесплодных людей. В этом отчете все пациенты с кариотипом 47, XYY бесплодны. Однако нельзя делать обобщений относительно корреляции между кариотипом и фенотипом бесплодия. Эта находка может быть случайной. Не было опубликовано систематических исследований, показывающих, что синдром XYY связан с повышенной частотой бесплодия. Опубликовано лишь несколько сообщений о случаях нарушений фертильности у мужчин с синдромом XYY [11]. Тем не менее, относительно высокая распространенность этого аномального кариотипа у бесплодных мужчин в нашем отчете оправдывает использование кариотипирования для оценки мужчин с репродуктивными аномалиями, особенно в случае мужчин с высокой степенью кровного родства.
Согласие
От пациентов было получено письменное информированное согласие на публикацию данного отчета.
Сокращения
- ФСГ:
фолликулостимулирующий гормон
- РЫБА:
флуоресцентная гибридизация in situ
- ГТГ:
G-диапазон с использованием трипсина и Гимзы
- Левая сторона:
лютеинизирующий гормон
Ссылки
Ratcliffe SG, Butler GE, James M: Эдинбургское исследование роста и развития детей с аномалиями половых хромосом. Дети и молодые люди с анеуплоидией половых хромосом. Под редакцией: Эванс Дж. А., Хамертон Дж. Л., Робинсон А. 1990, Нью-Йорк: Wiley-Liss for the National Foundation — March of Dimes, 59–115.
Google Scholar
Moorhead PS, Nowell PC, Mellman WJ, Battips DM, Hungerford D: Хромосомные препараты лейкоцитов, культивированных из периферической крови человека. Разрешение ячейки опыта. 1960, 20: 613-616. 10.1016/0014-4827(60)
Артикул КАС пабмед Google Scholar
ISCN 95, Мительман Ф.
(редактор): Международная система цитогенетической номенклатуры человека. 1995, Каргер, БазельGoogle Scholar
Ratcliffe SG, Read G, Pan H, Fear C, Lindenbaum R, Crossley J: Пренатальные уровни тестостерона у мужчин XXY и XYY. Горм Рез. 1994, 42: 106-109. 10.1159/000184157.
Артикул КАС пабмед Google Scholar
Foresta C, Moro E, Ferlin A: микроделеции Y-хромосомы и изменения сперматогенеза. Endocr Rev. 2001, 22: 226-239. 10.1210/er.22.2.226.
КАС пабмед Google Scholar
Крюгер Д.Г., Агерхольм И., Байриэль Л., Феддер Дж., Бруун-Петерсен Г.: Генетический анализ самцов из пар с интрацитоплазматической инъекцией сперматозоидов. Клин Жене. 2003, 64: 198-203. 10.1034/j.1399-0004.2003.00128.х.
Артикул КАС пабмед Google Scholar
Робинсон Д.О., Джейкобс П.А.: Происхождение дополнительной Y-хромосомы у мужчин с кариотипом 47,XYY. Хум Мол Жене. 1999, 8: 2205-2209. 10.1093/hmg/8.12.2205.
Артикул КАС пабмед Google Scholar
Morel F, Roux C, Bresson JL: Анеуплоидии половых хромосом в сперме 47 мужчин XYY. Арх Андрол. 1999, 43: 27-36. 10.1080/014850199262706.
Артикул КАС пабмед Google Scholar
Rives N, Milazzo JP, Miraaux L, North MO, Sibert L, Macé B: От сперматоцитов к сперматозоидам у бесплодного мужчины XYY. Int J Androl. 2005, 28: 304-310.
10.1111/j.1365-2605.2005.00540.х.Артикул пабмед Google Scholar
 «>
«>Martin RH: Цитогенетические детерминанты мужской фертильности. Хум Репрод. 2008, 14: 379-390. 10.1093/humupd/dmn017.
Артикул КАС Google Scholar
 (редактор): Международная система цитогенетической номенклатуры человека. 1995, Каргер, Базель
(редактор): Международная система цитогенетической номенклатуры человека. 1995, Каргер, Базель «>
«>Лилфорд Р., Джонс А.М., Бишоп Д.Т., Торнтон Дж., Мюллер Р.: Исследование «случай-контроль» о том, является ли бесплодие семейным. Бр Мед Дж. 1994, 309 (6954): 570-573.
Артикул КАС Google Scholar
 10.1111/j.1365-2605.2005.00540.х.
10.1111/j.1365-2605.2005.00540.х.Скачать ссылки
Благодарности
Авторы выражают благодарность Отделу генетики и репродукции человека медицинского факультета Университета Мансура, Египет, за техническую поддержку.
Информация об авторе
Авторы и организации
Отделение генетики и репродукции человека, медицинский факультет, Мансура, 35516, Египет
Фаеза Эль-Датори
Медицинский и ветеринарный университет Нотхемского университета , Кампус Саттон-Бонингтон, Лафборо, Лестершир, LE12 5RD, Великобритания
Hany M Elsheikha
Авторы
- Faeza El-Dahtory
Просмотр публикаций автора
Вы также можете искать этого автора в PubMed Google Scholar
- Hany M Elsheikha
Просмотр публикаций автора
Вы также можете искать этого автора в PubMed Google Scholar
Автор, ответственный за корреспонденцию

Дополнительная информация
Конкурирующие интересы
Авторы заявляют об отсутствии конкурирующих интересов.
Вклад авторов
FED провел цитогенетический анализ и сбор данных. ОН внес свой вклад в анализ, обзор литературы и написал статью. Оба автора прочитали и одобрили окончательный вариант рукописи.
Оригинальные файлы изображений, представленные авторами
Ниже приведены ссылки на оригинальные файлы изображений, представленные авторами.
Оригинальный файл авторов для рисунка 1
Оригинальный файл авторов для рисунка 2
Оригинальный файл авторов для рисунка 3
Права и разрешения
, Это статья в открытом доступе, распространяемая в соответствии с лицензией Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии, что оригинальная работа правильно цитируется.