ФГБНУ НЦПЗ. ‹‹Общая психиатрия››
Цитогенетика — это область генетики, связанная с изучением хромосом. Исследование хромосом при психической патологии показало, что диагностическое значение этот метод имеет в основном при умственной отсталости. При неврозах, эндогенных психозах, алкоголизме и других видах психической патологии хромосомные изменения, как правило, не обнаруживаются. Среди новорожденных частота хромосомной патологии равна примерно 0,6 %. Хромосомные болезни были известны еще до открытия самих хромосом (например, синдромы Клайнфельтера и Шерешевского — Тернера). Использование термина «болезнь» для хромосомной патологии не совсем верно, поскольку для любой болезни характерен тот или иной тип ее развития, т.е. закономерная смена симптомов и синдромов во времени. В случае же хромосомных аномалий совокупность специфических признаков больного — его фенотип является врожденным и практически не меняющимся. Большинство случаев хромосомных нарушений возникает спорадически в половых клетках здоровых родителей или на стадии первых делений зиготы.
Материалом для микроскопического исследования хромосом служат в основном лейкоциты крови, реже для этой цели используются культуры клеток кожи или костного мозга. Хромосомные аномалии могут возникать в половых и соматических клетках. Они представляют собой изменение числа хромосом — наличие добавочных хромосом или отсутствие хромосомы, или их перестройку, т.е. структурные изменения. Структурные изменения бывают внутрихромосомными — такими как делеция (утрата части хромосомы) или дупликация (удвоение участка хромосомы), а также межхромосомными — транслокация (обмен участками между хромосомами) и др.
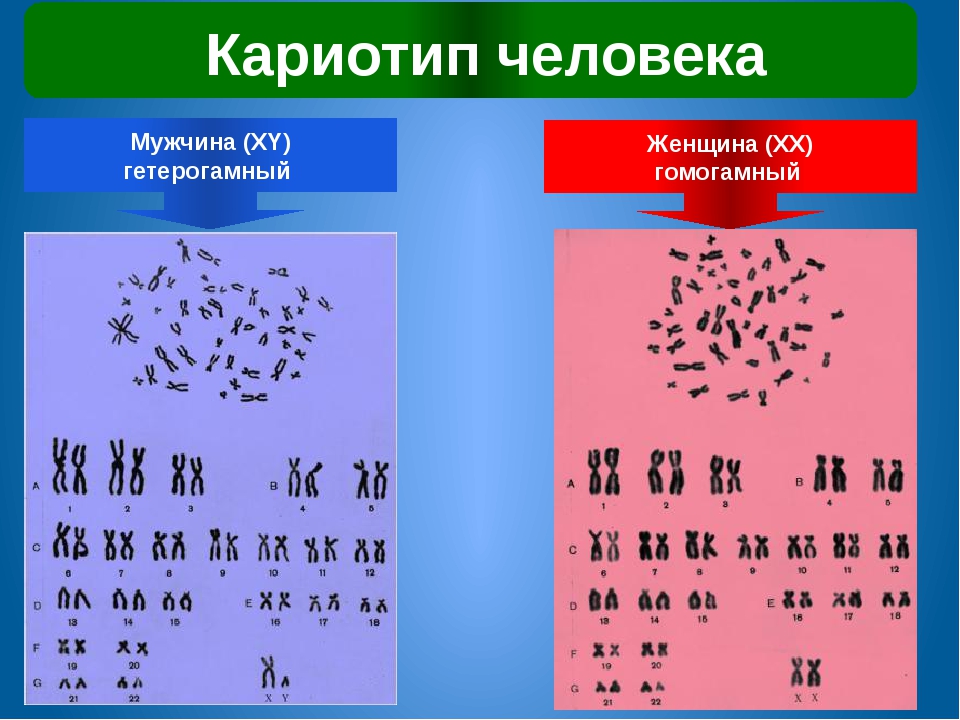
При описании кариотипа индивида указывают общее число хромосом, затем состав половых хромосом, наличие транслокации или мозаицизма и т.д. Например, запись 46XY означает нормальный мужской кариотип; 46ХХ — нормальный женский кариотип. Если указано 47XXY, то это кариотип синдрома Клайнфельтера, когда имеется дополнительная Х-хромосома. Кариотип 45X0 соответствует синдрому Шерешевского — Тернера, обусловленному отсутствием одной Х-хромосомы. Добавочная аутосомная хромосома указывается своим номером и знаком плюс, например, 47ХХ, 21+ обозначает кариотип девочки с лишней 21-й хромосомой (болезнь Дауна), а утрата хромосомы обозначается соответствующим номером и знаком минус. Транслокация обозначается буквой «t» с указанием номеров хромосом, которые обменялись участками, например 45XY, t(14+21). Наличие мозаичных клеток обозначается соответствующими кариотипами с использованием знака дроби, например 45X0/46, XX — мозаичность по синдрому Шерешевского — Тернера (45X0). При описании кариотипов используются также и другие символы, с правилами применения которых можно ознакомиться в специальных руководствах.
В случае хромосомной патологии почти все клинические проявления сопровождаются множественными нарушениями в строении тела и психики, причем их выраженность сильно варьирует при одних и тех же хромосомных аномалиях. Например, при болезни Дауна поражение психики проявляется слабоумием от легкой до тяжелой степени. Было также отмечено, что у больных с аномалиями аутосомных хромосом интеллект нарушается в большей степени, чем при аномалиях половых хромосом.
Цитогенетические исследования
Кариотип — это совокупность признаков полного набора хромосом соматических клеток организма на стадии метафазы (III фаза деления клетки) – их количество, размер, форма, особенности строения. Исследование кариотипа проводят методом световой микроскопии с целью выявления патологии хромосом. Чаще всего это исследование проводят у детей для выявления заболеваний, обусловленных нарушениями в хромосомах и у супругов при бесплодии, или привычном невынашивании беременности.
Нарушения нормального кариотипа у человека возникают на ранних стадиях эмбрионального развития организма. Если имеются нарушения кариотипа в половых клетках будущих родителей, то кариотип зиготы, образовавшейся при слиянии родительских клеток, также оказывается нарушенным.При дальнейшем делении такой зиготы все клетки эмбриона и развившегося из него организма окажутся с одинаково аномальным кариотипом.
Как правило, нарушения кариотипа у человека сопровождаются различными, в том числе комплексными, пороками развития, и большинство таких аномалий несовместимо с жизнью. Это приводит к самопроизвольным абортам на ранних стадиях беременности. Однако достаточно большое число плодов (~2,5%) с аномальными кариотипами донашивают до окончания беременности.
Цитогенетические методы исследования позволяют выявлять нарушения кариотипа — числа и структуры хромосом, хромосомной патологии. Такая патология может являться причиной мужского и женского бесплодия, а также тяжелых хромосомных заболеваний у детей (например синдрома Дауна).
Такая патология может являться причиной мужского и женского бесплодия, а также тяжелых хромосомных заболеваний у детей (например синдрома Дауна).
Кариотипирование рекомендовано всем супружеским парам при планировании беременности, в случае замерзшей беременности, невынашивании беременности.
Современные методы кариотипирования обеспечивают детальное обнаружение хромосомных аберраций (внутрихромосомных и межхромосомных перестроек), нарушения порядка расположения фрагментов хромосом — делеции, дупликации, инверсии, транслокации. Такое исследование кариотипа позволяет диагностировать ряд хромосомных заболеваний, вызванных как грубыми нарушениями кариотипов (нарушение числа хромосом), так и нарушением хромосомной структуры или множественностью клеточных кариотипов в организме.
- В лаборатории выполняется:
- Исследование кариотипа с раскладкой 11 ; 29 и 100 метафазных пластинок для исключения уровня вероятности мозаицизма от 25% до 3%. Для определения кариотипа достаточно исследовать 11 метафазных пластин.

- Исследование кариотипа для определения фрагильной (ломкой) Х- хромосомы (анализируется 100 метафазных пластин).
- Для идентичности описания результатов цитогенетических исследований используется Международная цитогенетическая номенклатура (International System for Cytogenetic Nomenclature, ISCN) и при обнаружении патологии дается подробная расшифровка полученного результата с точным описанием отдельных хромосом и их участков.

| График выдачи результатов молекулярно-биологисческих и цитогенетических исследовании |
|
ПЦР исследования на 2-4 рабочий день Молекулярно- генетические исследования выдаются по графику Цитогенетические исследования выполняются в течение 14-18 рабочих дней |
Назад
Анализ на кариотип супругов в «Надия Одесса»: высокая точность и эффективность
Когда мужчина и женщина вступает в брак, они мечтают о долгой и счастливой жизни и здоровых детках.
Это хромосомный набор человека, другими словами его генетический паспорт, неизменный на протяжении жизни. У человека в норме 46 хромосом, по 23 хромосомы от каждого родителя. Графически это выглядит так: нормальный женский кариотип – 46, ХХ и нормальный мужской – 46 ХY.
У человека может быть участок перестроенных хромосом и даже не знать об этом. Но о проблеме он узнает тогда, когда будет планировать беременность. Дефект хромосом становится причиной выкидыша, замершей беременности или ребёнка с генетическими болезнями.
Изменить кариотип человека невозможно, но если вовремя сдать кровь на кариотип и узнать причину бесплодия, то можно подобрать решение этой проблемы. Сегодня пары обращаются к искусственному оплодотворению, а также обращаются за помощью донорского материала – спермы или яйцеклетки.
Сегодня пары обращаются к искусственному оплодотворению, а также обращаются за помощью донорского материала – спермы или яйцеклетки.
- Невынашивание беременности;
- Первичное и вторичное бесплодие;
- Мертворождение или рождение ребёнка с врождёнными пороками;
- У родственников супругов есть хромосомные аномалии;
- В показателях спермы супруга имеются отклонения;
- Ранняя менопауза;
- Отсутствие менструации.
- Анализа на кариотип супругов: как правильно подготовиться? Анализ сдается натощак. За месяц до кариотипирования нужно отказаться от приёма антибиотиков, а за три дня не пить алкогольных напитков. Если один из супругов болел простудным заболеванием, то анализ нужно перенести на две недели
- Как происходит анализ? Чтобы определить кариотип, используются одноядерные лейкоциты или культуру клеток. Этот анализ имеет много непонятных для обычного человека формул и терминов, поэтому более подробно вы сможете это расспросить у своего врача.
- О чём ещё нужно знать? Обычно исследование кариотипа человека назначает врач-генетик. На практике используется один из видов кариотипирования: стандартное или молекулярное. Стандартное не может выявить большую часть хромосомных патологий, поэтому его назначают редко. Молекулярное кариотипирование позволяет определить большую часть патологий, но не все, поэтому генетик может назначить и другие исследование.
- Где сдать анализ на кариотип? Супруги могут сдать анализ на кариотип в специализированных центрах репродуктивной медицины. В клинике «Ivf-clinic» вы можете сделать кариотипирование и узнать результат. У нас работают репродуктологи высшей категории, а современная лаборатория оснащена высокотехнологичным оборудованием. Быстро и безопасно персонал клиники проведёт процедуру забора крови и отправит его в работу.
 Этот анализ имеет много непонятных для обычного человека формул и терминов, поэтому более подробно вы сможете это расспросить у своего врача.
Этот анализ имеет много непонятных для обычного человека формул и терминов, поэтому более подробно вы сможете это расспросить у своего врача.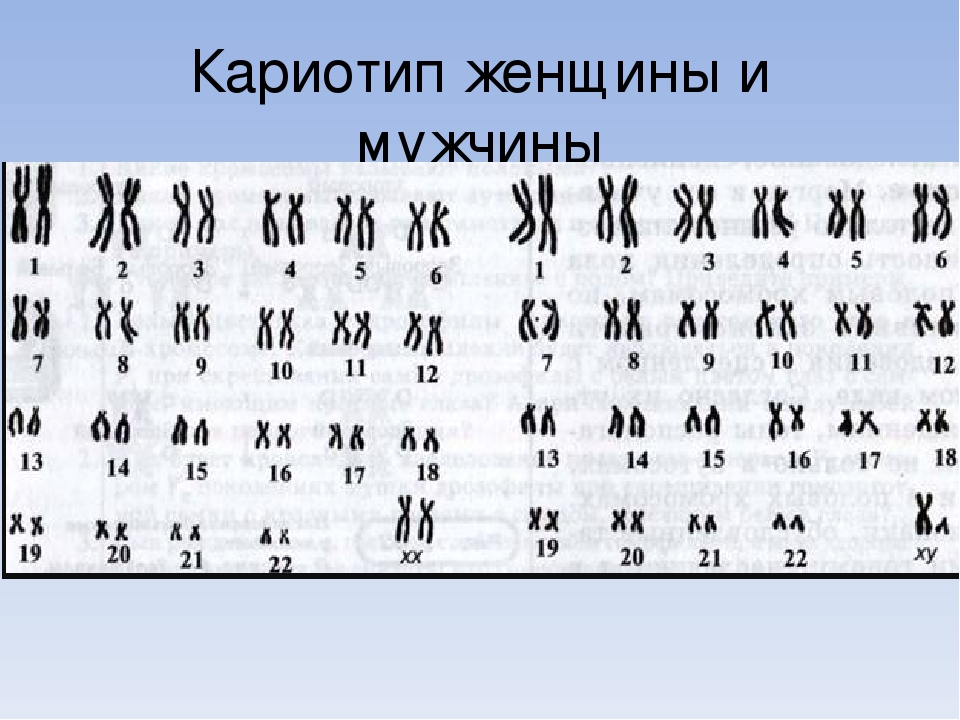
Запишитесь на приём в клинику по горячей линии 0 800 750-390.
Смотрите также:
Клиника ЭКО | Кариотипирование
Кариотипирование — генетическое исследование № 1 во всем мире. Это цитогенетическое исследование хромосомного набора человека (кариотипа), которое позволяет по анализу крови выявить изменения в количественном и качественном составе хромосом. Кариотип — это хромосомная характеристика вида, представленная определенным числом и морфологией хромосом, их пространственной организацией и распределением эу- и гетерохроматина. Кариотип человека в норме состоит из 46 хромосом (23 пары): 22 пары аутосом и 1 пара половых хромосом (ХХ или XY). Нормальный женский кариотип кратко обозначается как 46,ХХ, нормальный мужской кариотип — 46,ХY.
Хромосомная патология может никак не проявлять себя в вашей жизни. Для многих мужчин и женщин нарушения кариотипа становятся полной неожиданностью. Выявление патологии дает возможность преодолеть сложности, к которым она приводит.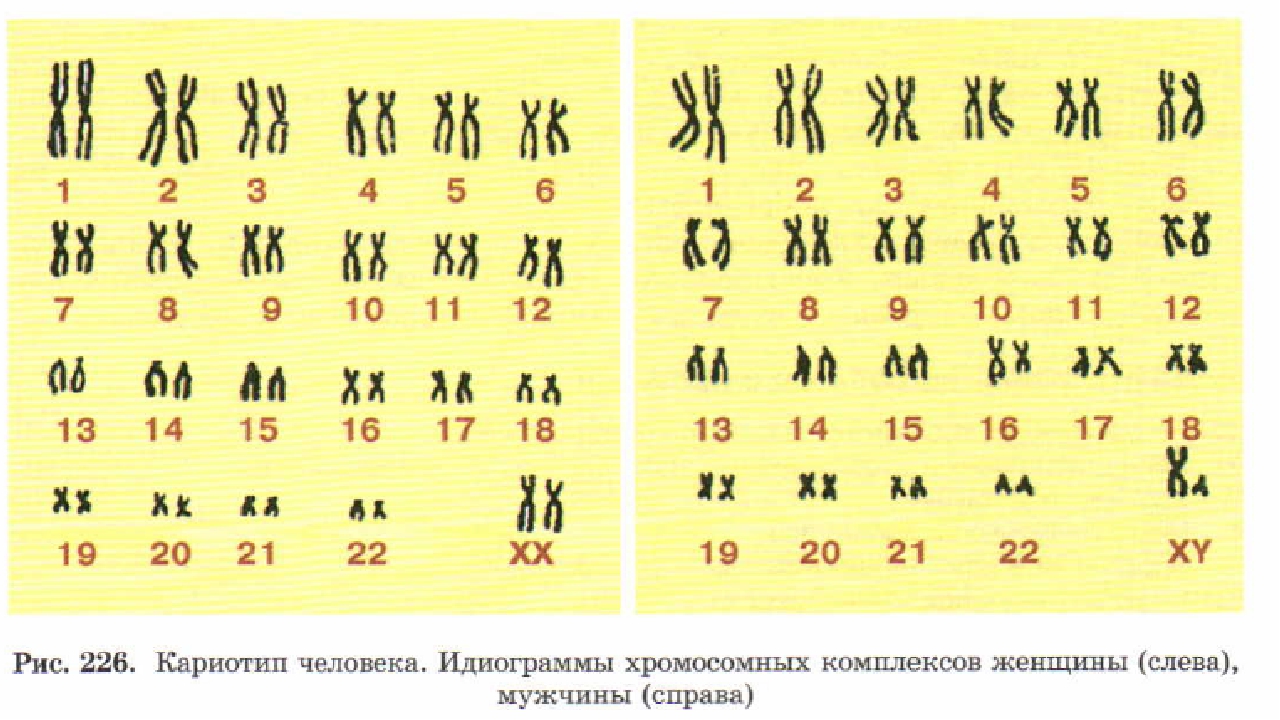
{vivod-form-priem}
Нарушения кариотипа могут приводить к следующим последствиям:
-
первичное бесплодие;
-
привычное невынашивание беременности;
-
рождение детей с патологией/пороками развития;
-
развитие генетически обусловленных заболеваний.
Показания к кариотипированию
Клиника МАМА рекомендует пройти исследование кариотипа всем мужчинам и женщинам, планирующим стать родителям. Вне зависимости от того, нуждаетесь ли вы в специализированном лечении или нет.
Пройдите кариотипирование, если:
-
у вас нарушение сперматогенеза;
-
в вашей семье есть наследственные заболевания;
-
у вас не получается выносить ребенка;
-
у вас бесплодие «неясного происхождения»;
-
лечение методом ЭКО не приносит результатов;
-
в вашей семье есть дети/родственники с врожденными заболеваниями/пороками развития.

Кариотипирование в Клинике МАМА
Кариотипирование — это хромосомный анализ лимфоцитов крови. В центре репродукции МАМА анализ кариотипа с аберрациями (внутрихромосомными и межхромосомными перестройками) проводят цитогенетическим методом с использованием сочетания способов дифференцированного и монохромного окрашивания хромосом. Это позволяет провести развернутое цитогенетическое исследование: определить число и структуру хромосом, выявить возможные перестройки — транслокации, инверсии, инсерции, дупликации, делеции.
Генетик Наталья Александровна Дорощук о кариотипе:
— Зачем сдавать этот анализ?
— Что делать, если обнаружены отклонения в анализе?
— Как планировать беременность при изменениях в кариотипе?
Результаты кариотипирования в Клинике МАМА
На основании проведенного исследования можно с высокой вероятностью выявить такие патологии, как:
-
делеция — утрата участка хромосомы;
-
дупликация — удвоение какого-либо фрагмента хромосомы;
-
инверсия — разворот участка хромосомы;
-
моносомия — в паре хромосом отсутствует одна хромосома;
-
транслокация — перемещение части одной хромосомы на другую;
-
трисомия — третья (лишняя) хромосома в паре.
-
Кариотипирование также позволяет оценить следующие риски:
-
нарушение имплантации эмбрионов;
-
невынашивание беременности;
-
возможность применения половых клеток (сперматозоиды, ооциты) пациента для ЭКО;

Кариотипирование — возможность узнать, что мешает вам забеременеть, выносить и родить здорового ребенка, а самое главное — успешно преодолеть это. Современная репродуктивная медицина позволяет предупреждать патологическую беременность благодаря методикам экстракорпорального оплодотворения и преимплантационной генетической диагностики.
Анализ на кариотип с аберрациями
Добрый день! Сегодня мы с вами поговорим о генетическом кариотипировании, как обещали в прошлом видео. И поговорим мы о кариотипировании с аберрациями.Что собой представляет, в принципе, хромосома? Хромосома – это спрессованные нити ДНК, несущие информацию о геноме человека. Конечно, хромосомный набор у каждого вида свой. У человека 46 хромосом (23 пары хромосом). Одна из них является половой. У женщины две Х хромосомы, и нормальный женский кариотип – 46, ХХ. У мужчины – ХY и, соответственно, нормальный мужской кариотип – 46, XY.
Конечно, хромосомный набор у каждого вида свой. У человека 46 хромосом (23 пары хромосом). Одна из них является половой. У женщины две Х хромосомы, и нормальный женский кариотип – 46, ХХ. У мужчины – ХY и, соответственно, нормальный мужской кариотип – 46, XY.
Что такое аберрация?
Аберрация – это изменение количества и структуры хромосом. Они могут носить регулярный характер, могут быть нерегулярными.
Регулярные аберрации, как правило, закладываются во время зачатия или сразу после него. Нерегулярные аберрации могут возникать в результате каких-то вредных воздействий: химиотерапия, радиация и так далее. Важно получить наиболее полную информацию о геноме человека, о каких-то возможных хромосомных аномалиях. С этой целью лучше, конечно, проводить полный анализ кариотипирование с аберрациями.
Кому назначается такой анализ?
• В тех ситуациях, когда были достаточно частые случаи невынашивания.Особенно, если мы говорим о невынашивании на малых сроках, в первом триместре беременности.
 Потому что мы уже говорили с вами в предыдущем ролике, что именно хромосомными аномалиями, именно генетическим сбоем в эмбрионах обусловлены эти остановки беременности.
Потому что мы уже говорили с вами в предыдущем ролике, что именно хромосомными аномалиями, именно генетическим сбоем в эмбрионах обусловлены эти остановки беременности. • Так же это может касаться семейных пар, у которых в течение многих лет отмечается бесплодие. Речь идет о 5 годах и больше.
• Те женщины, у которых уже были попытки ЭКО и эти попытки были неудачными.
Как проводится этот анализ?
У человека забирается кровь, выделяются лимфоциты и, с помощью специальных препаратов – митогенов — эти лимфоциты заставляют делиться. Деление происходит до тех пор, пока могут визуализировать хромосомы. На этом этапе деление останавливают, делают специальные мазки на стекла и проводят непосредственно исследование.Если говорить о простом кариотипировании, исследуется 12 – 15 клеток. Если говорить о кариотипировании с аберрациям, то исследуется около 100 клеток. Поэтому, из этого можно сделать вывод, что это исследование более информативное, более углубленное.
Как подготовиться к сдаче анализа?
Безусловно, накануне не следует употреблять алкогольные напитки, не стоит употреблять жирную пищу.
Обязательно нужно посмотреть на общее состояние своего здоровья – то есть, чтобы не было никакого дискомфорта, катаральных явлений. Естественно, когда речь идет о каких-то острых заболеваниях: ОРВИ, ОРЗ, или об обострениях каких-то хронических заболеваний — это тоже не совсем удачное время для того, чтобы проводить данный анализ.
В наших клиниках такие исследования проводятся довольно давно. Мы имеем большой опыт. Поэтому, если у вас есть какие-то вопросы, если вы хотите обсудить какие-то темы, особенно если вы живете далеко, не в Москве, пожалуйста, задавайте свои вопросы, напишите в комментариях. Мы с удовольствием вам ответим!
У нас есть возможность проведения онлайн-консультации для женщин, которые живут не в московском регионе. У нас есть возможность очных консультаций.
Пожалуйста, приходите. Мы всегда будем рады помочь вам!
Нарушения формирования пола — ДНК-диагностика
Инверсия пола, 46,XY
Наличие женского фенотипа при нормальном мужском кариотипе характеризует XY-инверсию пола.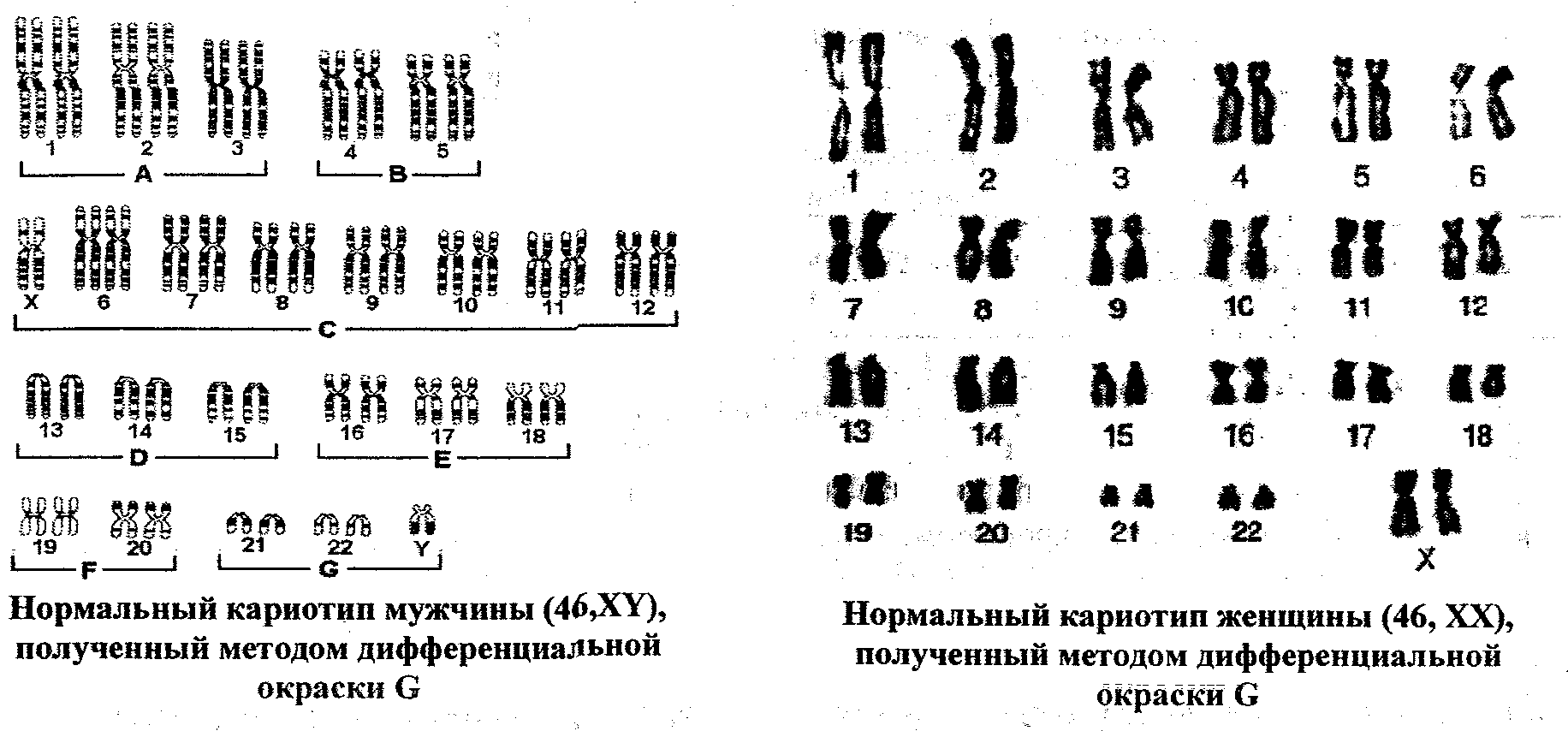 Наиболее частой причиной данного нарушения формирования пола является синдром Свайера – это полная или «чистая» дисгенезия гонад при кариотипе 46,XY. Частота XY-дисгенезии гонад составляет 1 на 30000 человек. Больные имеют женский фенотип без признаков двойственности полового развития: феминное телосложение, развитые по женскому типу наружные половые органы, нормально развитую или гипоплазированную матку и маточные (фаллопиевы) трубы. Однако у пациентов с синдромом Свайера практически отсутствуют женские половые железы, которые в данном случае представлены дисгенетичными гонадами, представляющими собой соединительнотканные тяжи (стреки) с небольшими включениями железистой ткани, овариально-подобной стромы без фолликулов. Как правило, диагностирование синдрома Свайера происходит у девочек в пубертатный период, когда у них не происходит нормального полового развития. Причиной обращения к врачу при этом является задержка полового развития и отсутствие начала менструаций, реже наличие злокачественных новообразований, происходящих из дисгенетичных гонад.
Наиболее частой причиной данного нарушения формирования пола является синдром Свайера – это полная или «чистая» дисгенезия гонад при кариотипе 46,XY. Частота XY-дисгенезии гонад составляет 1 на 30000 человек. Больные имеют женский фенотип без признаков двойственности полового развития: феминное телосложение, развитые по женскому типу наружные половые органы, нормально развитую или гипоплазированную матку и маточные (фаллопиевы) трубы. Однако у пациентов с синдромом Свайера практически отсутствуют женские половые железы, которые в данном случае представлены дисгенетичными гонадами, представляющими собой соединительнотканные тяжи (стреки) с небольшими включениями железистой ткани, овариально-подобной стромы без фолликулов. Как правило, диагностирование синдрома Свайера происходит у девочек в пубертатный период, когда у них не происходит нормального полового развития. Причиной обращения к врачу при этом является задержка полового развития и отсутствие начала менструаций, реже наличие злокачественных новообразований, происходящих из дисгенетичных гонад. Так как дисгенетичные гонады подвержены озлокачествлению, показано их удаление в детстве или на момент постановки диагноза XY-дисгенезии гонад. После оперативного лечения пациенткам, как правило, еще в подростковом возрасте назначается заместительная гормональная терапия, чтобы достичь нормального развития вторичных половых признаков и предотвратить развитие остеопороза. У женщин с XY-дисгенезией гонад нет собственных яйцеклеток, однако в некоторых случаях она в состоянии выносить плод, полученный в программе ЭКО при оплодотворении донорской яйцеклетки сперматозоидами супруга.
Так как дисгенетичные гонады подвержены озлокачествлению, показано их удаление в детстве или на момент постановки диагноза XY-дисгенезии гонад. После оперативного лечения пациенткам, как правило, еще в подростковом возрасте назначается заместительная гормональная терапия, чтобы достичь нормального развития вторичных половых признаков и предотвратить развитие остеопороза. У женщин с XY-дисгенезией гонад нет собственных яйцеклеток, однако в некоторых случаях она в состоянии выносить плод, полученный в программе ЭКО при оплодотворении донорской яйцеклетки сперматозоидами супруга.
Инверсия пола, 46,XY тип 1 (OMIM 400044)
Наиболее частой из известных причин «чистой» формы дисгенезии гонад 46,XY являются микроструктурные перестройки Y-хромосомы c утратой гена SRY (Sex-determining region Y), а также точковые мутации данного гена. У 10-15% больных с синдромом Свайера обнаруживают отсутствие локуса SRY. В большинстве случаев это обусловлено утратой фрагмента дистальной части короткого плеча Y-хромосомы (Yp11.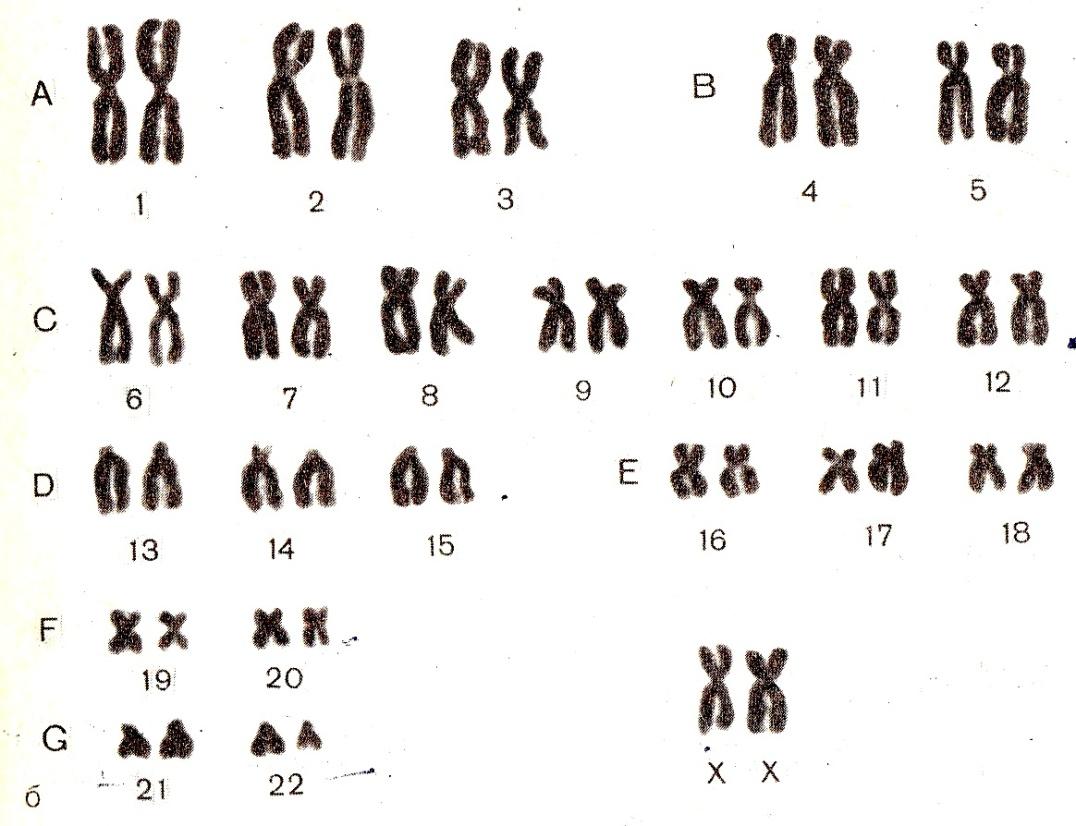 3), вследствие X-Y транслокации. Еще у 10-15% пациентов с данным синдромом выявляют мутации гена SRY.
3), вследствие X-Y транслокации. Еще у 10-15% пациентов с данным синдромом выявляют мутации гена SRY.
Ген SRY локализован на коротком плече Y хромосомы и кодирует транскрипционный фактор – белок, связывающийся с генами, определяющими развитие пола плода по мужскому типу. Мутации в гене SRY приводят к синтезу функционально неполноценного белка и к нарушению дифференцировки клеток Сертоли и формирования семенных канальцев в развивающихся бипотенциальных гонадах плода, что вызывает дисгенезию гонад и развитие остальных органов половой системы по женскому типу, несмотря на наличие Y-хромосомы в кариотипе.
Инверсия пола, 46,XYтип 2 (OMIM 300018)
Данный тип XY-инверсии пола обусловлен дупликаций гена NR0B1 (DAX-1). Ген NR0B1локализован на коротком плече Х хромосомы (локус Хp21.3). Кодируемый этим геном белок DAX-1 играет важную роль в развитии и функции некоторых органов эндокринной системы, в том числе и половых желез. Еще внутриутробно он контролирует активность генов, участвующих в формировании этих тканей, а в постнатальном периоде DAX-1 регулирует выработку в них гормонов. Белок DAX-1 оказывает дозо-зависимый эффект на органы эндокринной системы. Дупликация гена NR0B1, а также делеция располагающегося рядом с геном NR0B1 локуса, негативно-регулирующего его транскрипцию приводит к XY-инверсии пола, обусловленной XY-дисгенезией гонад часто сочетающейся с нарушением функции надпочечников. Точковые мутации этого гена у пациентов с кариотипом 46,XY вызывают нарушение развития тестикулярной ткани, приводят к дефициту маскулинизации. Мутации в этом гене также вызывают Х-сцепленную гипоплазию надпочечников, как у пациентов с кариотипом 46,ХХ так и 46,XY.
Белок DAX-1 оказывает дозо-зависимый эффект на органы эндокринной системы. Дупликация гена NR0B1, а также делеция располагающегося рядом с геном NR0B1 локуса, негативно-регулирующего его транскрипцию приводит к XY-инверсии пола, обусловленной XY-дисгенезией гонад часто сочетающейся с нарушением функции надпочечников. Точковые мутации этого гена у пациентов с кариотипом 46,XY вызывают нарушение развития тестикулярной ткани, приводят к дефициту маскулинизации. Мутации в этом гене также вызывают Х-сцепленную гипоплазию надпочечников, как у пациентов с кариотипом 46,ХХ так и 46,XY.
Инверсия пола, 46,XY тип 3 (OMIM 612965)
Данная форма XY-инверсии пола обусловлена мутациями гена NR5A1 (SF1). Ген NR5A1 кодирует транскрипционный фактор — стероидогенный фактор 1 (SF-1), с помощью которого контролируется активность ряда генов, кодирующих экспрессию белков-ферментов, ответственных за биосинтез стероидных гормонов в надпочечниках и гонадах, в том числе выработку половых гормонов. Функция белка SF-1 регулирует дифференцировку, развитие и функционирование, надпочечников, мужских и женских половых желез, сперматогенез и оогенез, развитие мужских или женских половых признаков. Ген NR5A1 локализуется на длинном плече хромосомы 9 (локус q33.3) и состоит из 7 экзонов (включая первый некодирующий экзон). Мутации в данном гене приводят к различным формам нарушения развития и функции половой и эндокринной систем. При этом нарушение дифференцировки и развития гонад, гаметогенеза может отмечаться как в сочетании, так и без поражения надпочечников (гипоплазия коры надпочечников). Помимо полной (характеризующейся наличием тяжевидных гонад при развитии остальных половых органов по женскому типу) и неполной формы (характеризующейся двойственным развитием гениталий) дисгенезии гонад 46,XY, мутации в гене NR5A1 могут приводить к развитию других заболеваний. Среди них: различная степень нарушения развития яичников у женщин с кариотипом 46,ХХ (от полной формы ХХ-дисгенезии гонад, неполной формы ХХ-дисгенезии гонад до синдрома преждевременной недостаточности яичников), развитие недостаточности коры надпочечников, синдрома тестикулярной дисгенезии и/или с нарушением сперматогенеза у 46,XY мужчин, бесплодие.
Функция белка SF-1 регулирует дифференцировку, развитие и функционирование, надпочечников, мужских и женских половых желез, сперматогенез и оогенез, развитие мужских или женских половых признаков. Ген NR5A1 локализуется на длинном плече хромосомы 9 (локус q33.3) и состоит из 7 экзонов (включая первый некодирующий экзон). Мутации в данном гене приводят к различным формам нарушения развития и функции половой и эндокринной систем. При этом нарушение дифференцировки и развития гонад, гаметогенеза может отмечаться как в сочетании, так и без поражения надпочечников (гипоплазия коры надпочечников). Помимо полной (характеризующейся наличием тяжевидных гонад при развитии остальных половых органов по женскому типу) и неполной формы (характеризующейся двойственным развитием гениталий) дисгенезии гонад 46,XY, мутации в гене NR5A1 могут приводить к развитию других заболеваний. Среди них: различная степень нарушения развития яичников у женщин с кариотипом 46,ХХ (от полной формы ХХ-дисгенезии гонад, неполной формы ХХ-дисгенезии гонад до синдрома преждевременной недостаточности яичников), развитие недостаточности коры надпочечников, синдрома тестикулярной дисгенезии и/или с нарушением сперматогенеза у 46,XY мужчин, бесплодие.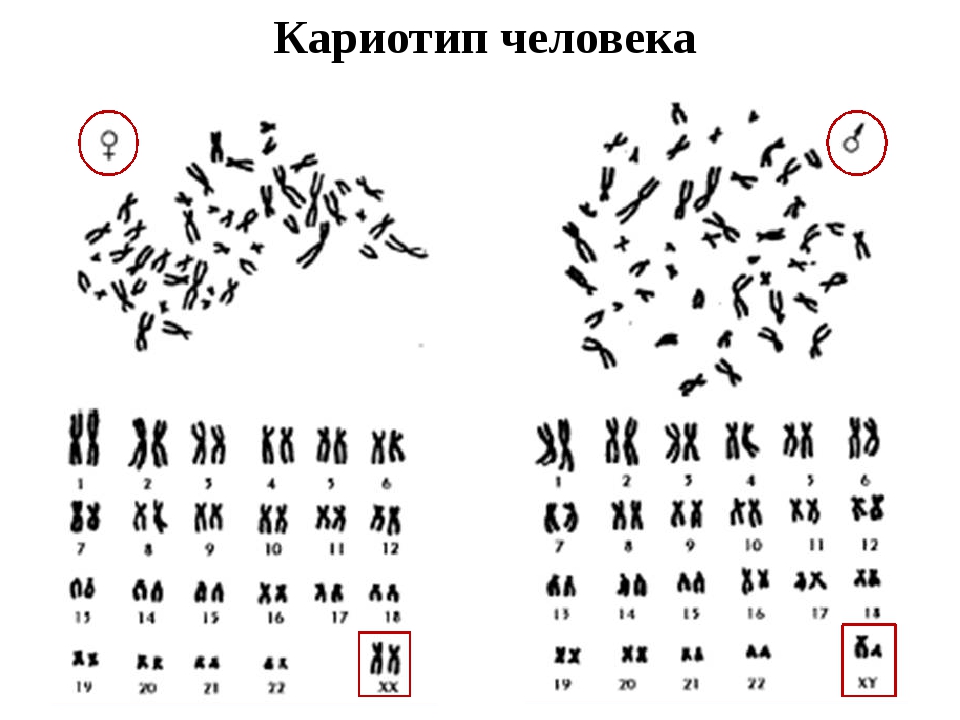 Исследование на наличие герминальных мутаций в гене NR5A1 показано при различных нарушениях формирования пола, полового развития или репродуктивной функции, соматические мутации – при опухолях надпочечников. Важно отметить, что нарушения, вызванные мутациями данного гена, в отличие от других генов, могут иметь как аутосомно-доминантный, так и аутосомно-рецессивный тип наследования. При этом у пациентов, имеющих мутации гена NR5A1, может быть, как нормальный мужской 46,XY, так и нормальный женский 46,ХХ кариотип.
Исследование на наличие герминальных мутаций в гене NR5A1 показано при различных нарушениях формирования пола, полового развития или репродуктивной функции, соматические мутации – при опухолях надпочечников. Важно отметить, что нарушения, вызванные мутациями данного гена, в отличие от других генов, могут иметь как аутосомно-доминантный, так и аутосомно-рецессивный тип наследования. При этом у пациентов, имеющих мутации гена NR5A1, может быть, как нормальный мужской 46,XY, так и нормальный женский 46,ХХ кариотип.
Инверсия пола, 46,XY тип 4 (OMIM 154230)
Эта форма XY-инверсии пола обусловлена делецией локуса 9p24.3. У пациенток отмечают нормально развитые по женскому типу наружные половые органы, нормально развитую или гипоплазированную матку, при гистологическом исследовании гонад обнаруживают наличие незрелой тестикулярной ткани, содержащей клетки Сертолли, и отсутствие зрелых половых клеток. Инверсия пола у данных пациентов, вероятно, обусловлена потерей одной из копий дозо-чувствительного гена, локализованного в данном локусе. Генами-кандидатами являются DMRT1 и DMRT2.
Генами-кандидатами являются DMRT1 и DMRT2.
Инверсия пола, 46,XY тип 5 (OMIM 613080)
Данная аутосомно-рецессивная форма инверсии 46,XY обусловлена наличием мутаций в гене CBX2, расположенного на хромосоме 17 (локус 17q25). В 2009 году Байсон-Лаубер описал случай новорожденной девочки с кариотипом 46,XY, у которой в результате секвенированияв гене CBX2 были обнаружены две мутации (P98L и R443P). В результате исследований у девочки были обнаружены нормально развитые яичники, с наличием овариальной ткани и первичных фолликулов, а также влагалище и матка. Однако возраст еще был слишком мал, чтобы оценить ее фертильность и дальнейшее половое развитие.
Инверсия пола, 46,XY тип 6 (OMIM 613762)
XY-инверсия пола связана с наличием мутации в гетерозиготном состоянии в гене MAP3K1, расположенном в локусе 5q11.2. Пациентки с данной формой дисгенезии гонад имеют высокий рост, который, вероятно, обусловлен избыточной продукцией андрогенов, тяжевидные яичники, гипоплазированную матку, иногда наблюдается клиторомегалия.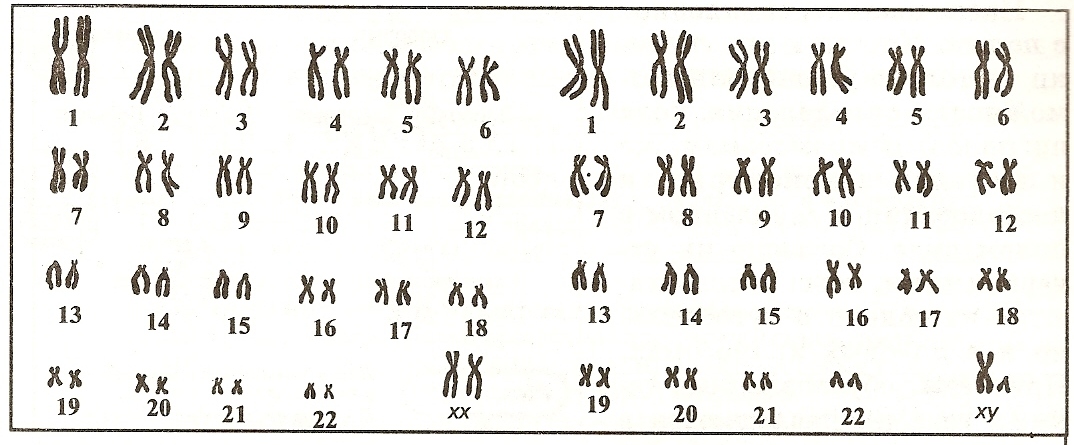
Инверсия пола, 46,XY тип 7 (OMIM 233420)
Инверсия пола обусловлена наличием у пациенток мутаций в гомозиготном или компаунд-гетерозиготном состоянии в гене DHH, расположенного в локусе 12q13.12. У нескольких пациенток было описано наличие недоразвитой матки, также присутствовали фаллопиевы трубы и наблюдали полную форму ХY-дисгенезии гонад (тяжевидные гонады, которые часто озлокачествлялись).
Инверсия пола, 46,XYтип 8 (OMIM 614279)
Данный тип XY-инверсии пола обусловлен мутациями гена AKR1C2, лежащего в локусе 10p15, отвечающего за альтернативный путь синтеза дигидротестостерона. Мутации сцепленного с ним гена AKR1C4, который сегрегирует вместе с геном AKR1C2, могут влиять на выраженность фенотипических проявлений.
В Центре Молекулярной Генетики проводится молекулярный анализ ключевых генов, контролирующих дифференцировку пола, в частности выполняется секвенирование генов SRY и NR5A1 (SF1), а также с помощью количественного метода MLPA проводится поиск делеций и дупликаций генов SRY, NR5A1 (SF1), NR0B1 (DAX-1).
Нами разработан набор для определения пола и регистрации нарушений детерминации пола на основе детекции генов SRY, AMG, AMGL и определения резус-фактора методом ПЦР. Набор предназначен для использования в диагностических лабораториях молекулярно-генетического профиля.
Публикации по теме раздела
Инверсия пола 46 XYИнформация о кариологическом исследовании и его применении
Это исследование совмещает качественный анализ (изучение внешнего строения) и количественный анализ (подсчет) хромосом. Хромосомы – это обязательно присутствующие в клетке особые компоненты, несущие информацию о наследственности. Хромосомы состоят из молекулы ДНК (основного носителя наследственной информации) и белков.
Обычно в соматических (неполовых) клетках человека (в клетках крови, кожи и пр.) содержится 46 хромосом, 23 пары: 23 – материнского происхождения, 23 – отцовского. Нормальный женский кариотип: 46,XX. Кариотип мужчины в норме: 46,XY.
Кариотип мужчины в норме: 46,XY.
Большая часть пороков развития, а также эндокринных, неврологических, гематологических заболеваний и проблем репродуктивного характера связана либо с изменением количества хромосом (геномные мутации), либо со структурными перестройками (хромосомные мутации).
Геномные мутации связаны с изменением количественного состава хромосомного набора (кариотипа). Наличие в клетках дополнительной хромосомы, как и отсутствие одной из хромосом, всегда влечет за собой серьезные проблемы для здоровья и нормального развития. Например, болезнь Дауна – присутствие в кариотипе дополнительной (третьей) хромосомы 21 – ведет к отставанию в умственном развитии, возникновению проблем с физическим здоровьем. Кариотип такого больного чаще всего выглядит так: 47,XX (или XY),+21. Отсутствие у девочки одной Х-хромосомы влечет задержку роста и полового развития и называется синдромом Тернера (45,X).
Хромосомные мутации – нарушение структуры одной или более хромосом с сохранением их нормального числа (46). Структурные перестройки вызываются разрывами в основе хромосомы – ДНК. Иногда происходит утрата части материала, удвоение, обмен участками между хромосомами. Если разные хромосомы обменялись между собой материалом без утраты его части, то говорят о сбалансированной транслокации. Такое изменение чаще всего на здоровье носителя (человека, у которого выявлена транслокация) не влияет, но может спровоцировать патологию у его будущего ребенка.
Структурные перестройки вызываются разрывами в основе хромосомы – ДНК. Иногда происходит утрата части материала, удвоение, обмен участками между хромосомами. Если разные хромосомы обменялись между собой материалом без утраты его части, то говорят о сбалансированной транслокации. Такое изменение чаще всего на здоровье носителя (человека, у которого выявлена транслокация) не влияет, но может спровоцировать патологию у его будущего ребенка.
Хромосомные нарушения имеют разную природу. Это или врожденное изменение кариотипа клеток всего организма, или приобретенное изменение хромосом отдельных клеток (например, при гематологических заболеваниях).
Самойлова Людмила Руслановна,
врач-лаборант-цитогенетик
Синдром Свайера: MedlinePlus Genetics
Синдром Свайера — это состояние, которое влияет на половое развитие. Половое развитие обычно идет по определенному пути, основанному на хромосомах человека; однако при синдроме Свайера половое развитие не типично для хромосомного паттерна пораженного человека.
У людей обычно 46 хромосом в каждой клетке. Две из 46 хромосом, известные как X и Y, называются половыми хромосомами, потому что они помогают определить, будут ли у человека развиваться мужские или женские половые признаки.Девочки и женщины обычно имеют две Х-хромосомы (кариотип 46,ХХ), тогда как мальчики и мужчины обычно имеют одну Х-хромосому и одну Y-хромосому (кариотип 46,ХУ). При синдроме Свайера у людей есть одна X-хромосома и одна Y-хромосома в каждой клетке, что обычно встречается у мальчиков и мужчин; однако у них есть женские репродуктивные структуры.
Люди с синдромом Свайера имеют женские наружные гениталии и некоторые женские внутренние гениталии; матка и фаллопиевы трубы нормально сформированы, но гонады (яичники или яички) не функционируют.Вместо этого гонады маленькие, недоразвитые и содержат мало гонадной ткани. Эти структуры называются полосатыми гонадами. Полоса ткани гонады подвержена риску развития рака, который трудно обнаружить, поэтому ее обычно удаляют хирургическим путем. Синдром Свайера иногда называют полной дисгенезией половых желез 46,XY; медицинский термин «дисгенезия» указывает на то, что развитие (при этом состоянии развитие гонад) снижено и нетипично.
Синдром Свайера иногда называют полной дисгенезией половых желез 46,XY; медицинский термин «дисгенезия» указывает на то, что развитие (при этом состоянии развитие гонад) снижено и нетипично.
Люди с синдромом Свайера обычно воспитываются как девочки и имеют женскую гендерную идентичность.Синдром Свайера может быть выявлен до рождения, при рождении или позже, когда у ребенка не происходит полового созревания, как обычно. Поскольку у них нет функциональных яичников, вырабатывающих гормоны, больные люди часто начинают заместительную гормональную терапию в подростковом возрасте, чтобы начать половое созревание, вызывая рост груди и матки и, в конечном итоге, приводя к менструации. Заместительная гормональная терапия также стимулирует развитие костей и помогает снизить риск аномально низкой плотности костей (остеопения и остеопороз).У женщин с синдромом Свайера не образуются яйцеклетки (яйцеклетки), но они могут забеременеть с помощью донорской яйцеклетки или эмбриона.
Всегда ли кариотип 46XX женский?
BMJ Case Rep. 2012; 2012: bcr2012006223.
2012; 2012: bcr2012006223.
Редкое заболевание
Ayesha AHMAD
Ayesha AHMAD
1
1 Алигарх Мусульманский университет, Алигарх, Уттар Прадеш, Индия
Mohammad ASIM Siddiqui
2 Департамент эндокринологии, Индивидуальная больница Аполлона, Нью-Дели, Индия
Anju Goyal
2 Отделение эндокринологии, больница Индрапрастха Аполлон, Нью-Дели, Индия
Субхаш Кумар Вангноо
2 Отделение эндокринологии, больница Индрапрастха Аполлон, Нью-Дели, Индия, Алитарх Муслим
0 Университет Алитарх, Алитарх 1 Прадеш, Индия
2 Отделение эндокринологии, больница Индрапрастха Аполло, Нью-Дели, Индия
Эта статья цитировалась в других статьях PMC.Abstract
19-летний мужчина из страны Ближнего Востока был направлен врачом в эндокринное отделение по поводу двусторонней гинекомастии, низкого либидо и редких волос на лице. В анамнезе не было никаких хронических заболеваний, эпидемического паротита или травматических повреждений яичка. У него были клинические признаки, указывающие на дефицит гонадотропина, который был подтвержден при биохимическом тестировании. При анализе кариотипа и флуоресцентной гибридизации in situ у него был обнаружен кариотип 46XX(SRY+).
В анамнезе не было никаких хронических заболеваний, эпидемического паротита или травматических повреждений яичка. У него были клинические признаки, указывающие на дефицит гонадотропина, который был подтвержден при биохимическом тестировании. При анализе кариотипа и флуоресцентной гибридизации in situ у него был обнаружен кариотип 46XX(SRY+).
Исходная информация
Тестикулярное нарушение полового развития 46XX характеризуется наличием кариотипа 46XX, мужскими наружными гениталиями в диапазоне от нормального до неоднозначного, двумя яичками, азооспермией и отсутствием мюллеровых структур.Большинство людей с тестикулярным расстройством половой дифференцировки (DSD) 46XX после полового созревания имеют нормальные лобковые волосы и нормальный размер полового члена, но маленькие яички, гинекомастию и бесплодие в результате азооспермии. Меньший процент имеет неоднозначные гениталии при рождении, когда это можно распознать на ранней стадии. Гендерная роль и гендерная идентичность указаны как мужские. Если не выявить и не лечить, мужчины с тестикулярным DSD 46XX испытывают последствия дефицита тестостерона.
Если не выявить и не лечить, мужчины с тестикулярным DSD 46XX испытывают последствия дефицита тестостерона.
Описание случая
19-летний мужчина из ближневосточной страны был направлен врачом в эндокринное отделение по поводу двусторонней гинекомастии и редких волос на лице (). Он был третьим из шести братьев и сестер, состоявших в кровнородственном браке, и имел мужские наружные гениталии. У него был нормальный антенатальный анамнез, и вехи его развития были нормальными. В анамнезе не было никаких признаков приема запрещенных наркотиков, хронических заболеваний, эпидемического паротита или травматических повреждений яичек. При дальнейшем допросе он жаловался на снижение либидо и снижение частоты бритья.Рост пациента составлял 165 см, волосы в подмышечных впадинах и на лобке нормальные. Его голос был нормальным. Его IQ был 100. Длина полового члена в растянутом состоянии составляла 7 см, но оба яичка были мягкими, маленькими и атрофичными (). Кожа была нормальной, пальпируемых нервов или миотонии не было. Клинических признаков тиреотоксикоза, заболеваний печени или почек или дефицита питательных веществ не было.
Клинических признаков тиреотоксикоза, заболеваний печени или почек или дефицита питательных веществ не было.
Пациент с гинекомастией.
Нормальная длина полового члена и уменьшенный объем яичек по данным орхидометра.
Исследования
Заметно повышены уровни гонадотропинов: фолликулостимулирующего гормона (ФСГ) — 15 мМЕ/мл и лютинизирующего гормона (ЛГ) — 20 мМЕ/мл (норма: ФСГ 1,2–5 мМЕ/мл; ЛГ 2–9,8). мМЕ/мл). Тестостерон в сыворотке был низким, но уровни пролактина и профиль щитовидной железы были в пределах нормы. МРТ малого таза не выявила наличия мюллеровых структур или каких-либо аномалий мочевыделительной системы. Ультрасонограмма показала правое яичко размером 21×15×10 мм и левое яичко размером 23×17×12 мм.Анализ спермы выявил азооспермию, объем спермы составил 3 мл, она была положительной на фруктозу. Двухэнергетическая рентгеновская абсорбциометрия выявила остеопению. Уровни витамина D были 16 нг/мл (что свидетельствует о дефиците). Хромосомный анализ периферической крови с использованием 72 ч. Стимулированная культура-GTG (предварительная обработка трипсином, затем окраска по Гимзе) (предварительная обработка трипсином, затем окраска по Гимзе), полоса, которая показала хромосомный набор 46XX (). Анализ флуоресцентной гибридизации in situ (FISH) проводили с использованием центромерных зондов для хромосом X и Y (Vysis CEPX/CEPY), чтобы исключить мозаицизм низкого уровня, и зонда, специфичного для области, определяющей пол (SRY) (Vysis CEPX/LSI SRY). для выявления любой скрытой перестройки, приводящей к транслокации области SRY на хромосому, отличную от Y, или для анализа присутствия области SRY даже в отсутствие Y-хромосомы.Все 500 клеток и 20 метафаз выявили два «зеленых» сигнала для «Х» хромосомы и один «оранжевый» сигнал, указывающий на присутствие области SRY на одной из «Х» хромосом, что согласуется с диагнозом 46XX (SRY+) ().
Стимулированная культура-GTG (предварительная обработка трипсином, затем окраска по Гимзе) (предварительная обработка трипсином, затем окраска по Гимзе), полоса, которая показала хромосомный набор 46XX (). Анализ флуоресцентной гибридизации in situ (FISH) проводили с использованием центромерных зондов для хромосом X и Y (Vysis CEPX/CEPY), чтобы исключить мозаицизм низкого уровня, и зонда, специфичного для области, определяющей пол (SRY) (Vysis CEPX/LSI SRY). для выявления любой скрытой перестройки, приводящей к транслокации области SRY на хромосому, отличную от Y, или для анализа присутствия области SRY даже в отсутствие Y-хромосомы.Все 500 клеток и 20 метафаз выявили два «зеленых» сигнала для «Х» хромосомы и один «оранжевый» сигнал, указывающий на присутствие области SRY на одной из «Х» хромосом, что согласуется с диагнозом 46XX (SRY+) ().
Хромосомный анализ периферической крови с использованием 72 ч. Стимулированная культура-GTG полоса, показывающая хромосомный набор 46XX.
FISH демонстрирует два «зеленых» сигнала для «X»-хромосомы и один «оранжевый» сигнал, указывающий на присутствие области SRY на одной из «X»-хромосом, что соответствует диагнозу 46XX (SRY+).
Дифференциальный диагноз
Синдром Клайнфельтера (47XXY) и его варианты (48XXXY, 49XXXXY и мозаицизм 46XY/47XXY) подозревают у мужчин с гипогонадизмом, маленькими яичками и гинекомастией.
46XX/46XY — могут представлять собой истинных гермафродитов в зависимости от относительного соотношения клеток XX и XY: фенотип может варьироваться от нормального мужчины до нормальной женщины.
45X/46XY — пораженные лица часто проявляются как мужчины и могут иметь низкий рост в зависимости от процентного содержания клеток 45X.
Синдромное XX расстройство половой дифференцировки яичек (DSD) — характеризуется ладонно-подошвенным кератозом и предрасположенностью к плоскоклеточному раку кожи. Было показано, что они связаны с мутацией R-спондина 1 (RSPO1).
46XX DSD яичек может быть связан с микрофтальмией и линейными дефектами кожи, когда аномальный обмен X/Y включает микроделецию Xp.
46XX овотестикулярный DSD — это настоящие гермафродиты (имеют ткань как яичка, так и яичника либо в виде яйцеклетки, либо в виде яичника и яичка), тогда как люди с 46XX тестикулярной DSD имеют только ткань яичка. Возможная систематическая ошибка при отборе проб биопсии гонад может пропустить овариальную часть гонад. Матка или полуматка могут быть продемонстрированы рентгенологически, а эндокринная оценка может выявить выработку эстрогена.
Возможная систематическая ошибка при отборе проб биопсии гонад может пропустить овариальную часть гонад. Матка или полуматка могут быть продемонстрированы рентгенологически, а эндокринная оценка может выявить выработку эстрогена.
Лечение
Ему была начата заместительная терапия низкими дозами тестостерона, а затем полная заместительная терапия. Также проводилась заместительная терапия кальцием и витамином D. Поскольку его больше беспокоила гинекомастия, ему посоветовали редукционную маммопластику, если заместительная терапия тестостероном не привела к улучшению.
Консультирование проводилось лечащей эндокринной бригадой и психологом для пациента и его родителей (с его согласия). Им подробно рассказали о диагнозе и о том, что маловероятно, что он станет отцом/детями. Были обсуждены варианты вспомогательной репродукции, и им было предоставлено право выбора в соответствии с их социокультурными, религиозными и личными предпочтениями.
Исход и наблюдение
Так как пациент был не из нашей страны, ему было представлено подробное резюме с возможностью постоянной консультации на родине. Разъяснено, что необходимо периодически проводить клиническое обследование, костную денситометрию, определение размера простаты и простатспецифического антигена, исследование функции печени. Если возможно, ему было предложено вернуться для проверки через 6 месяцев.
Разъяснено, что необходимо периодически проводить клиническое обследование, костную денситометрию, определение размера простаты и простатспецифического антигена, исследование функции печени. Если возможно, ему было предложено вернуться для проверки через 6 месяцев.
Обсуждение
Мужской синдром XX был впервые описан de la Chapelle et al. 1 в 1964 году, встречается примерно у 1 из 20 000 новорожденных мальчиков. Фенотипически выделяют три группы особей 46XX с инверсией пола. В первую классическую группу входят фенотипически нормальные мужчины XX, во вторую группу входят мужчины с генитальной двойственностью, а в третью группу входят истинные гермафродиты. 2
В 1966 году Ferguson-Smith 3 и его коллеги предположили, что это состояние возникает в результате транслокации материала Y, включая SRY, с Y-хромосомы на X-хромосому. McElreavey et al 4 предположили, что SRY постулируется для подавления ингибитора развития мужских яичек. SRY отрицательно регулирует аутосомный локус Z, который выключает гены, определяющие мужской пол. У нормальных мужчин SRY, расположенный на коротком плече Y-хромосомы непосредственно проксимальнее псевдоаутосомной области I, негативно регулирует Z-локус.У мужчин 46XX отсутствует длинное плечо Y-хромосомы (Yq), которое содержит локусы фактора азооспермии и комплекс генов, критический для развития и дифференцировки зародышевых клеток. Уровни ФСГ и ЛГ в данном случае были повышены, как это наблюдалось в случаях с тяжелой тестикулопатией и полной остановкой сперматогенеза, приводящей к тестикулярной недостаточности. Лица с 46XX (SRY+) редко имеют атипичные гениталии и реже, чем люди с 46XX (SRY-), имеют гинекомастию. 5
SRY отрицательно регулирует аутосомный локус Z, который выключает гены, определяющие мужской пол. У нормальных мужчин SRY, расположенный на коротком плече Y-хромосомы непосредственно проксимальнее псевдоаутосомной области I, негативно регулирует Z-локус.У мужчин 46XX отсутствует длинное плечо Y-хромосомы (Yq), которое содержит локусы фактора азооспермии и комплекс генов, критический для развития и дифференцировки зародышевых клеток. Уровни ФСГ и ЛГ в данном случае были повышены, как это наблюдалось в случаях с тяжелой тестикулопатией и полной остановкой сперматогенеза, приводящей к тестикулярной недостаточности. Лица с 46XX (SRY+) редко имеют атипичные гениталии и реже, чем люди с 46XX (SRY-), имеют гинекомастию. 5
Таким образом, для полного обследования мужчин XX важно, чтобы за обычным цитогенетическим анализом следовал анализ FISH для определения наличия гена SRY.Эти исследования показывают, что количество Y-специфического материала способствует гетерогенности фенотипа.
Очки обучения
Подробный анамнез, охватывающий все детали, необходим в тех случаях, когда фенотип не соответствует внешнему виду.
Постояльцы, не имеющие опыта работы с такими случаями, не должны чувствовать себя подавленными, когда сталкиваются с такими случаями.
После подтверждения диагноза пациенту, а также родителям/опекунам необходимо провести подробное психосоциальное консультирование с деликатным подходом, при этом все медицинские термины должны быть подробно объяснены.
Сноски
Конкурирующие интересы: Нет.
Согласие пациента: Получено.
Ссылки
1. de la Chapelle A, Hortling H, Niemi M, et al. ХХ половые хромосомы у человека мужского пола. Первый случай. Акта Мед Сканд 1964; 175 (Приложение 412): 25–8. [PubMed] [Google Scholar]2. Boucekkine C, Toublanc JE, Abbas N, et al. Клинический и анатомический спектр у пациентов XX с измененным полом. Отношение к наличию Y-специфических последовательностей ДНК.Клин Эндокринол (Oxf)
1994;40:733–42. [PubMed] [Google Scholar]3. Фергюсон-Смит, Массачусетс.
X-Y хромосомный обмен в этиологии истинного гермафродитизма и XX синдрома Клайнфельтера. Ланцет
1966; 2: 475–6. [PubMed] [Google Scholar]4. McElreavey K, Rappaport R, Vilain E, et al.
Меньшая часть из 46, XX настоящих гермафродитов положительна на последовательность Y-ДНК, включая SRY. Хум Жене
1992; 90: 121–5. [PubMed] [Google Scholar]5. Эргун-Лонгмайр Б., Винчи Г., Алонсо Л. и др.
Клиническая, гормональная и цитогенетическая оценка мужчин 46, XX и обзор литературы.J Педиатр Эндокринол Метаб
2005; 18: 739–48. [PubMed] [Google Scholar]
Отношение к наличию Y-специфических последовательностей ДНК.Клин Эндокринол (Oxf)
1994;40:733–42. [PubMed] [Google Scholar]3. Фергюсон-Смит, Массачусетс.
X-Y хромосомный обмен в этиологии истинного гермафродитизма и XX синдрома Клайнфельтера. Ланцет
1966; 2: 475–6. [PubMed] [Google Scholar]4. McElreavey K, Rappaport R, Vilain E, et al.
Меньшая часть из 46, XX настоящих гермафродитов положительна на последовательность Y-ДНК, включая SRY. Хум Жене
1992; 90: 121–5. [PubMed] [Google Scholar]5. Эргун-Лонгмайр Б., Винчи Г., Алонсо Л. и др.
Клиническая, гормональная и цитогенетическая оценка мужчин 46, XX и обзор литературы.J Педиатр Эндокринол Метаб
2005; 18: 739–48. [PubMed] [Google Scholar]Кариотип 46,XX — обзор
1.2.3 Аномалии развития гонад
У женщин с кариотипом 46,XX отсутствие развития гонад не приводит к каким-либо очевидным клиническим признакам. И внутренние, и внешние половые органы развиты по-женски. Большинство пациентов обращают на себя внимание из-за отсутствия полового развития из-за недостаточности половых желез в подростковом возрасте, и в это время медицинское обследование выявляет гипергонадотропный гипогонадизм. Были выявлены ассоциации с различными генами, поэтому следует искать точный диагноз. 35,36
Были выявлены ассоциации с различными генами, поэтому следует искать точный диагноз. 35,36
В некоторых очень редких случаях у людей с кариотипом 46,ХХ может запускаться развитие яичек. 37,38 В основном это вызвано транслокацией гена SRY в другую хромосому, но другие нижележащие гены также могут быть аномально отрегулированы, чтобы начать развитие яичек. В этих случаях часто присутствуют нормальные внутренние и внешние мужские гениталии, а эндокринная функция яичек остается нормальной.Однако мужчины 46,XX обычно бесплодны, так как зародышевые клетки дегенерировали. В классификации DSD это называется 46,XX тестикулярной DSD.
В некоторых случаях при кариотипе 46,ХХ развитие яичек частичное, что приводит к дифференцировке овотестикулярных гонад. 39 В этих случаях либо одна гонада дифференцируется как семенник, а другая как яичник, но в основном одна сторона содержит ткани как яичка, так и яичника, а другая сторона будет яичником. В зависимости от количества тестикулярной ткани и эндокринной активности у детей может быть вариабельное развитие матки, часто проявляющееся в виде полуматки на стороне яичников.Наружные половые органы могут быть частично вирилизованы в зависимости от уровня и активности тестостерона. Эта форма DSD называется овотестикулярной DSD, и она может проявляться как с кариотипом 46,XX, так и с 46,XY, хотя последний встречается гораздо реже. 40
В зависимости от количества тестикулярной ткани и эндокринной активности у детей может быть вариабельное развитие матки, часто проявляющееся в виде полуматки на стороне яичников.Наружные половые органы могут быть частично вирилизованы в зависимости от уровня и активности тестостерона. Эта форма DSD называется овотестикулярной DSD, и она может проявляться как с кариотипом 46,XX, так и с 46,XY, хотя последний встречается гораздо реже. 40
Чаще может наблюдаться дисгенезия гонад 46,XY. 41 У этих пациентов был описан широкий клинический спектр, обычно называемый частичной или полной дисгенезией гонад. При полной дисгенезии гонад 46,XY фенотип является полностью женским, и его нельзя отличить от дисгенезии гонад 46,XX.Женщины имеют нормально сформированную матку и генитальную анатомию, как у женщин. Поэтому в подростковом возрасте эти больные вновь обратятся в поле зрения из-за отсутствия полового развития и будет диагностирован гипергонадотропный гипогонадизм. Важно определить кариотип только для возможности развития опухоли из остатков гонад и выявить возможные сопутствующие клинические признаки.
Важно определить кариотип только для возможности развития опухоли из остатков гонад и выявить возможные сопутствующие клинические признаки.
Генетические причины дисгенезии гонад 46,XY разнообразны и часто еще не выяснены.Одной из причин может быть делеция или вредная мутация SRY -гена. Также описано мутаций SOX9 и мутаций в других генах каскада развития яичек. 42 Важное значение имеет обнаружение мутаций в гене опухоли Вильмса 1, так как этот ген также участвует в развитии почек. 43 У пациентов с полной дисгенезией гонад 46,XY, также называемой синдромом Фрейзера, развивается прогрессирующая почечная недостаточность, и им обычно требуется диализ в возрасте до 20 лет.Также у них высок риск развития опухолей гонад, преимущественно дисгермином, в раннем возрасте. Но и другие генетические причины дисгенезии гонад 46,XY могут иметь сопутствующие заболевания. У пациентов с мутациями SOX9 описано костное проявление, называемое кампомелической дисплазией. Мутации Desert Hedge Hog связаны с сенсорной полинейропатией и т. д. 44 Поэтому для пациента важно выяснить генетическую основу дисгенезии половых желез 46,XY.
Мутации Desert Hedge Hog связаны с сенсорной полинейропатией и т. д. 44 Поэтому для пациента важно выяснить генетическую основу дисгенезии половых желез 46,XY.
Частичная дисгенезия половых желез 46,XY является основным дифференциальным диагнозом при ДСД. 40,41 У этих детей наблюдается частичная андрогенизация наружных половых органов с высокой вариабельностью. Кроме того, оценка внутренних половых органов может выявить вариабельное развитие матки и фаллопиевых труб. Это связано с уменьшением секреции АМГ из частично дисгенетических яичек. Однако в широком спектре пациентов с частичной дисгенезией гонад у некоторых пациентов есть мюллеровы структуры, которые не видны макроскопически, но могут быть видны при гистологическом исследовании.Фенотипический спектр настолько изменчив, что некоторые дети однозначно воспитываются как девочки, тогда как другие, даже с мутациями в одном и том же гене, могут быть отнесены к мужскому полу. Другие могут быть откровенно двусмысленными. Генетические причины столь же широко распространены, как и при полной дисгенезии гонад. Совсем недавно было описано довольно много пациентов с мутациями NR5A1 , приводящими к дефициту SF-1. 45 Корреляции генотип-фенотип не существует. Редко описывается дополнительная надпочечниковая недостаточность. 46
Генетические причины столь же широко распространены, как и при полной дисгенезии гонад. Совсем недавно было описано довольно много пациентов с мутациями NR5A1 , приводящими к дефициту SF-1. 45 Корреляции генотип-фенотип не существует. Редко описывается дополнительная надпочечниковая недостаточность. 46
Другие гены связаны с дисгенезией гонад и также участвуют в генезе других органов.
Кариотип 46,XY — обзор
45,X/46,XY Мозаичность и варианты
Мозаичный кариотип 45,X/46,XY, иногда называемый смешанной дисгенезией гонад , вероятно, возникает из-за задержки анафазы во время митоз в зиготе, хотя иногда наблюдаются аномалии Y-хромосомы, а межхромосомные перестройки с потерей структурно-аномального Y-материала могут быть распространенным механизмом для вариантов этого состояния.Хотя классическая форма этого состояния связана с мозаицизмом 45,X/46,XY, также сообщалось о мозаичных кариотипах 45,X/47,XYY или 45,X/46,XY/47,XYY.
Клинический фенотип, связанный с мозаицизмом 45,X/46,XY, сильно варьирует, и истинная распространенность этого состояния неизвестна (см. Таблицу 23-4). Исторически лица с наиболее тяжелыми формами мозаицизма 45,X/46,XY направлялись для дальнейшего обследования, и большинство серий пациентов, описанных в литературе, вероятно, отражали это смещение, поскольку исследования, основанные на невыбранном пренатальном кариотипировании, показали, что большинство дети с мозаицизмом 45,X/46,XY выглядят как мальчики. 209-211
Зарегистрированные генитальные фенотипы, связанные с мозаицизмом 45,X/46,XY, варьируются от женских наружных гениталий или легкой клиторомегалии до всех стадий неоднозначных гениталий до гипоспадии или нормального полового члена. 209,212-215 Фенотипы гонад варьируют от полосатых гонад через дисгенетические яички до яичек с нормальной гистологической структурой. В редких случаях могут присутствовать яичникоподобная строма и редкие примордиальные фолликулы. Гонады могут располагаться в любом месте по пути опускания яичка, при этом полосатые гонады чаще располагаются внутри брюшной полости, а хорошо сформированные яички чаще располагаются в пахово-мошоночной области.Мюллеровы структуры могут присутствовать в наиболее тяжелых случаях из-за нарушения продукции АМГ клетками Сертоли. Заметные различия в развитии гонад и гистологическом виде можно увидеть между правой и левой сторонами или даже в пределах одной гонады (отсюда термин смешанная дисгенезия гонад ), что часто приводит к асимметрии наружных половых органов. 215 Наличие полуматки и маточной трубы на стороне наиболее сильно пораженной гонады в некоторых случаях является важным доказательством паракринного действия АМГ на развивающиеся мюллеровы структуры.
Гонады могут располагаться в любом месте по пути опускания яичка, при этом полосатые гонады чаще располагаются внутри брюшной полости, а хорошо сформированные яички чаще располагаются в пахово-мошоночной области.Мюллеровы структуры могут присутствовать в наиболее тяжелых случаях из-за нарушения продукции АМГ клетками Сертоли. Заметные различия в развитии гонад и гистологическом виде можно увидеть между правой и левой сторонами или даже в пределах одной гонады (отсюда термин смешанная дисгенезия гонад ), что часто приводит к асимметрии наружных половых органов. 215 Наличие полуматки и маточной трубы на стороне наиболее сильно пораженной гонады в некоторых случаях является важным доказательством паракринного действия АМГ на развивающиеся мюллеровы структуры.
Соматические признаки, связанные с кариотипом 45,X/46,XY, сильно варьируют и не всегда хорошо коррелируют с гонадным фенотипом. 209,213-215 Приблизительно 40% детей имеют дополнительные клинические признаки, напоминающие синдром Тернера, такие как низкий рост, затылочные складки, низко посаженная линия роста волос, сердечные и почечные аномалии. 215 Может потребоваться детальное обследование и длительное наблюдение за такими пациентами, как и при синдроме Тернера (см. главу 10).В других случаях уменьшение прогнозируемого роста может быть единственным соматическим проявлением. Для этой группы лиц может потребоваться постоянный мониторинг признаков, связанных с синдромом Тернера (например, функции щитовидной железы, слуха, сердечных аномалий), и многие семьи получают пользу от психологической поддержки и обучения, которые могут быть связаны со специализированными услугами.
215 Может потребоваться детальное обследование и длительное наблюдение за такими пациентами, как и при синдроме Тернера (см. главу 10).В других случаях уменьшение прогнозируемого роста может быть единственным соматическим проявлением. Для этой группы лиц может потребоваться постоянный мониторинг признаков, связанных с синдромом Тернера (например, функции щитовидной железы, слуха, сердечных аномалий), и многие семьи получают пользу от психологической поддержки и обучения, которые могут быть связаны со специализированными услугами.
Установление пола может быть затруднено у лиц с 45,X/46,XY, и следует учитывать несколько факторов, включая внешний вид гениталий и анатомию мочеполовой системы, риск злокачественного новообразования гонад, фертильность и репродуктивные возможности, потенциальную потребность в заместительной гормональной терапии и возможную гендерная идентичность, половое ролевое поведение и психосексуальное функционирование.
Большинство младенцев с женскими или минимально андрогенизированными гениталиями воспитываются как девочки, а наличие матки или половины матки допускает возможность беременности в результате донорства яйцеклеток в будущем, хотя прогнозирование будущей функции может быть затруднено.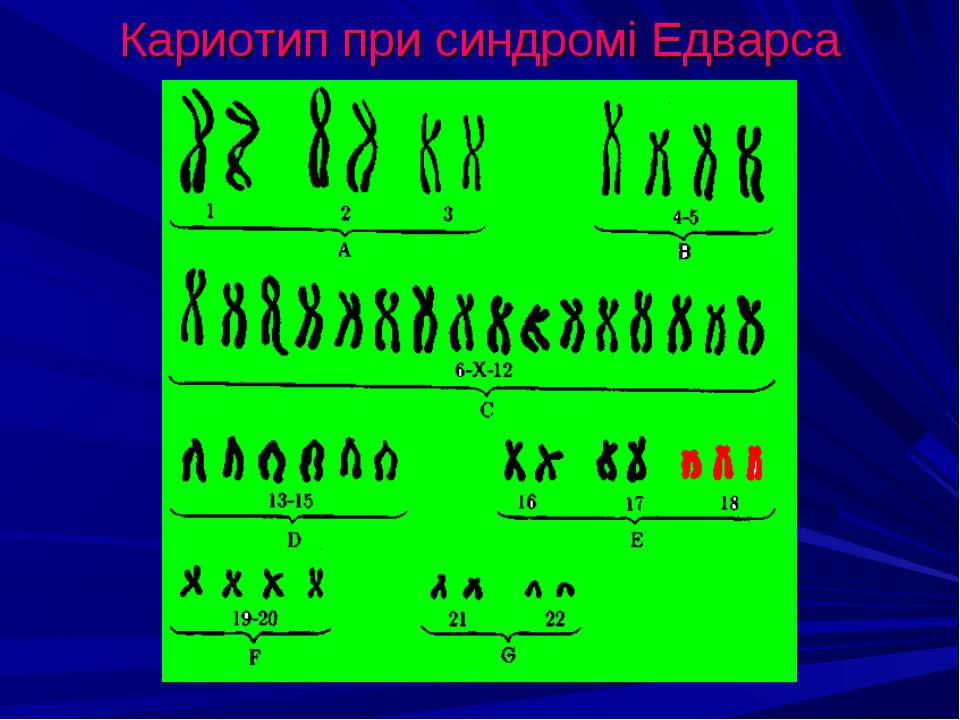 Считается, что внутрибрюшная полоса и дисгенетические гонады представляют значительный риск злокачественного новообразования и должны быть удалены из-за более высокого риска появления герминогенных опухолей в дисгенетических структурах. 212,214,216 Замена эстрогена необходима для стимуляции развития молочных желез и матки в подростковом возрасте, а добавление прогестинов способствует менструации при наличии матки.Стимуляторы роста использовались в индивидуальном порядке при наличии признаков низкого роста или синдрома Тернера. Крупных исследований для оценки этой группы пациентов не проводилось, хотя может наблюдаться значительная потеря роста в период полового созревания, и некоторые исследования предполагают, что использование ГР в раннем детстве может оптимизировать потенциал роста, если это вызывает беспокойство. 213,214,217 Точно так же отсутствуют данные о долгосрочных исходах в отношении гендерной идентичности или психосексуального функционирования.
Считается, что внутрибрюшная полоса и дисгенетические гонады представляют значительный риск злокачественного новообразования и должны быть удалены из-за более высокого риска появления герминогенных опухолей в дисгенетических структурах. 212,214,216 Замена эстрогена необходима для стимуляции развития молочных желез и матки в подростковом возрасте, а добавление прогестинов способствует менструации при наличии матки.Стимуляторы роста использовались в индивидуальном порядке при наличии признаков низкого роста или синдрома Тернера. Крупных исследований для оценки этой группы пациентов не проводилось, хотя может наблюдаться значительная потеря роста в период полового созревания, и некоторые исследования предполагают, что использование ГР в раннем детстве может оптимизировать потенциал роста, если это вызывает беспокойство. 213,214,217 Точно так же отсутствуют данные о долгосрочных исходах в отношении гендерной идентичности или психосексуального функционирования.
Младенцы с гипоспадией и нормальным фаллическим развитием обычно воспитываются как мальчики. Тестостерон иногда можно назначать для стимулирования роста фаллоса в младенчестве, а лечение гипоспадии обычно предлагается в виде двухэтапной процедуры. Следует попытаться выполнить орхидопексию как одно- или двухэтапную процедуру, поскольку может существовать значительный риск малигнизации этих гонад, и необходим тщательный мониторинг яичек. Половые железы, которые не могут быть помещены в мошонку, обычно удаляют. 212,214 Половые железы, которые можно зафиксировать в мошонке, нуждаются в тщательном наблюдении с помощью пальпации и биопсии в подростковом возрасте для выявления карциномы in situ. Некоторые исследования предполагают, что регулярное УЗИ яичек в подростковом возрасте можно использовать для выявления таких изменений, как микролитиаз. 212,214 Необходимо тщательно контролировать период полового созревания, чтобы обеспечить адекватную выработку эндогенного тестостерона, а в некоторых случаях необходимо дополнительное введение тестостерона. Большинство мальчиков вступают в период полового созревания спонтанно, но у некоторых затем развивается андрогенная недостаточность, и, как правило, они подвергаются урогенитальной хирургии.
Тестостерон иногда можно назначать для стимулирования роста фаллоса в младенчестве, а лечение гипоспадии обычно предлагается в виде двухэтапной процедуры. Следует попытаться выполнить орхидопексию как одно- или двухэтапную процедуру, поскольку может существовать значительный риск малигнизации этих гонад, и необходим тщательный мониторинг яичек. Половые железы, которые не могут быть помещены в мошонку, обычно удаляют. 212,214 Половые железы, которые можно зафиксировать в мошонке, нуждаются в тщательном наблюдении с помощью пальпации и биопсии в подростковом возрасте для выявления карциномы in situ. Некоторые исследования предполагают, что регулярное УЗИ яичек в подростковом возрасте можно использовать для выявления таких изменений, как микролитиаз. 212,214 Необходимо тщательно контролировать период полового созревания, чтобы обеспечить адекватную выработку эндогенного тестостерона, а в некоторых случаях необходимо дополнительное введение тестостерона. Большинство мальчиков вступают в период полового созревания спонтанно, но у некоторых затем развивается андрогенная недостаточность, и, как правило, они подвергаются урогенитальной хирургии. 214 Уменьшение конечного роста неизменно, но у некоторых мальчиков наблюдается положительный ответ на гормон роста. 213,214,217
214 Уменьшение конечного роста неизменно, но у некоторых мальчиков наблюдается положительный ответ на гормон роста. 213,214,217
Назначение пола и ведение ребенка 45,X/46,XY с весьма неоднозначными гениталиями может быть трудной ситуацией для родителей и врачей, и данные о долгосрочных исходах для этой группы недоступны. Ограниченные данные свидетельствуют о том, что примерно 60% младенцев с этим фенотипом воспитываются женского пола, но они бесплодны, у них нет матки, требуется гонадэктомия и, вероятно, будет проведена урогенитальная операция.Напротив, те, кто вырос как мужчина, часто подвергаются множественным операциям по поводу гипоспадии, могут иметь плохую корпоральную ткань, быть бесплодными, если присутствуют дисгенетические гонады, которые необходимо удалить, и могут иметь значительно сниженный потенциал роста. Детальная оценка каждого ребенка важна, а индивидуальный подход со стороны опытной междисциплинарной команды важен для управления и долгосрочного наблюдения и поддержки. Данные о долгосрочных исходах из более крупных исследований могут дать лучшее руководство по ведению этой группы людей в будущем.
Данные о долгосрочных исходах из более крупных исследований могут дать лучшее руководство по ведению этой группы людей в будущем.
В дополнение к наиболее тяжелым случаям, описанным ранее, мозаичный кариотип 45,X/46,XY может быть связан с мужским фенотипом и, по-видимому, нормальным развитием яичек. Первоначальные случаи нормальных мужчин с кариотипом 45,X/46,XY были описаны после скрининга членов семьи как потенциальных доноров трансплантации костного мозга, но более поздние исследования амниоцентеза показали, что 90% плодов с диагнозом 45,X/46,XY амниоцентез и подтвержденный наличие этого кариотипа имеют нормальные мужские гениталии и, по-видимому, нормальные яички после рождения. 210,211 Более того, существует ограниченная корреляция между степенью мозаицизма в образцах периферической крови и гонадным или соматическим фенотипом. Данные последующего наблюдения в этой когорте ограничены, функция HPG подробно не сообщалась, а исходы фертильности и риск развития опухолей неизвестны. Хотя кариотип 45,X/46,XY является редкой находкой у мужчин с опухолями яичка или в клинике бесплодия, необходимы более подробные долгосрочные исследования когорты мужчин 45,X/46,XY, чтобы узнать, будут ли обширные наблюдения — вверх необходимо.Может показаться разумным контролировать функцию гонад в этой когорте и оценивать признаки карциномы яичка in situ в подростковом возрасте, но доказательства для наилучшего подхода отсутствуют.
Хотя кариотип 45,X/46,XY является редкой находкой у мужчин с опухолями яичка или в клинике бесплодия, необходимы более подробные долгосрочные исследования когорты мужчин 45,X/46,XY, чтобы узнать, будут ли обширные наблюдения — вверх необходимо.Может показаться разумным контролировать функцию гонад в этой когорте и оценивать признаки карциномы яичка in situ в подростковом возрасте, но доказательства для наилучшего подхода отсутствуют.
13.1C: Идентификация хромосом и кариотипов
Кариотип показывает количество, размер и любые аномалии хромосом в организме.
Идентификация хромосом
Выделение и микроскопическое исследование хромосом составляет основу цитогенетики и является основным методом, с помощью которого клиницисты выявляют хромосомные аномалии у людей.Кариотип – это количество и внешний вид хромосом. Чтобы получить представление о кариотипе человека, цитологи фотографируют хромосомы, а затем вырезают и вставляют каждую хромосому в диаграмму или кариограмму, также известную как идеограмма.
У данного вида хромосомы можно идентифицировать по их количеству, размеру, положению центромер и характеру полос. В кариотипе человека аутосомы или «хромосомы тела» (все неполовые хромосомы) обычно расположены примерно в порядке размера от наибольшего (хромосома 1) до наименьшего (хромосома 22).Однако на самом деле хромосома 21 короче хромосомы 22. Это было обнаружено после того, как синдром Дауна был назван трисомией 21, что отражает то, как это заболевание возникает из-за наличия одной дополнительной хромосомы 21 (всего три). Не желая менять название этого важного заболевания, хромосома 21 сохранила свою нумерацию, несмотря на описание самого короткого набора хромосом. Хромосомы X и Y не являются аутосомами и называются половыми хромосомами.
Хромосомные «плечи», выступающие с любого конца центромеры, могут быть обозначены как короткие или длинные, в зависимости от их относительной длины.Короткое плечо обозначается аббревиатурой p («маленький»), тогда как длинное плечо обозначается аббревиатурой q (потому что оно следует за буквой «p» в алфавитном порядке).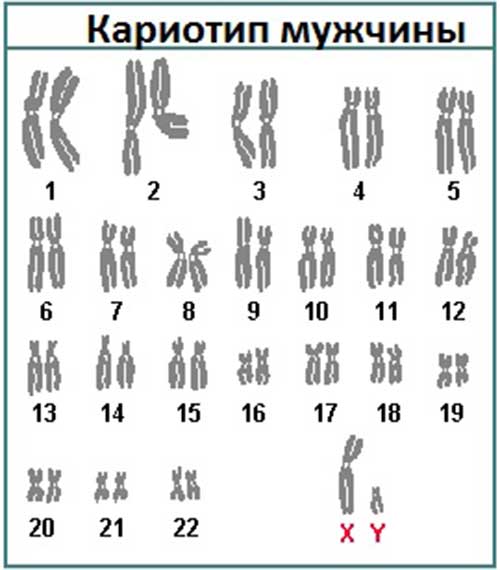 Каждая рука дополнительно подразделяется и обозначается номером. Используя эту систему именования, местоположения на хромосомах могут быть последовательно описаны в научной литературе.
Каждая рука дополнительно подразделяется и обозначается номером. Используя эту систему именования, местоположения на хромосомах могут быть последовательно описаны в научной литературе.
Хотя Менделя называют «отцом современной генетики», он проводил свои эксперименты, не используя ни одного из инструментов, которые обычно используют современные генетики. Одним из таких мощных цитологических методов является кариотипирование, метод, при котором признаки, характеризующиеся хромосомными аномалиями, могут быть идентифицированы в одной клетке.Чтобы определить кариотип человека, его клетки (например, лейкоциты) сначала берут из образца крови или другой ткани. В лаборатории изолированные клетки стимулируют к активному делению. Затем к клеткам применяется химическое вещество под названием колхицин, чтобы остановить конденсированные хромосомы в метафазе. Затем клетки набухают с помощью гипотонического раствора, чтобы хромосомы расходились. Наконец, образец сохраняется в фиксаторе и наносится на предметное стекло.
Затем генетик окрашивает хромосомы одним из нескольких красителей, чтобы лучше визуализировать отчетливые и воспроизводимые образцы полос каждой пары хромосом.После окрашивания хромосомы просматривают с помощью светлопольной микроскопии. Распространенным выбором окраски является окраска по Гимзе. Окрашивание по Гимзе дает примерно 400–800 полос (плотно спиральной ДНК и конденсированных белков), расположенных вдоль всех 23 пар хромосом. Опытный генетик может идентифицировать каждую хромосому на основе ее характерного рисунка полос. В дополнение к образцам полос хромосомы дополнительно идентифицируются на основе размера и расположения центромер. Чтобы получить классическое изображение кариотипа, в котором гомологичные пары хромосом выровнены в порядке номеров от самой длинной до самой короткой, генетик получает цифровое изображение, идентифицирует каждую хромосому и вручную упорядочивает хромосомы в этот образец.
Рисунок \(\PageIndex{1}\): Кариотип человека : Этот кариотип принадлежит мужчине. Обратите внимание, что гомологичные хромосомы имеют одинаковый размер, одинаковое положение центромер и структуру полос. Человеческая женщина будет иметь пару хромосом XX вместо показанной пары XY.
Обратите внимание, что гомологичные хромосомы имеют одинаковый размер, одинаковое положение центромер и структуру полос. Человеческая женщина будет иметь пару хромосом XX вместо показанной пары XY. По сути, кариотип может выявить генетические аномалии, при которых у человека слишком много или слишком мало хромосом на клетку. Примерами этого являются синдром Дауна, который идентифицируется по третьей копии хромосомы 21, и синдром Тернера, который характеризуется наличием у женщин только одной Х-хромосомы вместо нормальных двух.Генетики также могут идентифицировать большие делеции или вставки ДНК. Например, синдром Якобсена, который включает в себя отличительные черты лица, а также пороки сердца и кровотечения, идентифицируется делецией на хромосоме 11. Наконец, кариотип может точно определить транслокации, которые происходят, когда сегмент генетического материала отрывается от одной хромосомы и снова прикрепляется. на другую хромосому или на другую часть той же хромосомы. Транслокации связаны с некоторыми видами рака, включая хронический миелогенный лейкоз.
При жизни Менделя наследование было абстрактным понятием, которое можно было вывести только путем проведения скрещиваний и наблюдения за признаками, проявляемыми потомством. Наблюдая за кариотипом, современные генетики могут фактически визуализировать хромосомный состав человека, чтобы подтвердить или предсказать генетические аномалии у потомства еще до рождения.
ЛИЦЕНЗИИ И СВИДЕТЕЛЬСТВА
CC ЛИЦЕНЗИОННЫЙ КОНТЕНТ, РАСПРОСТРАНЕННЫЙ РАНЕЕ
- Курирование и доработка. Предоставлено : Boundless.com. Лицензия : CC BY-SA: Attribution-ShareAlike
CC ЛИЦЕНЗИОННОЕ СОДЕРЖИМОЕ, КОНКРЕТНОЕ АВТОРСТВО
- гемизиготные. Предоставлено : Викисловарь. Расположен по адресу : http://en.wiktionary.org/wiki/hemizygous . Лицензия : CC BY-SA: Attribution-ShareAlike
- Колледж OpenStax, биология. 16 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44481/latest…ol11448/latest . Лицензия : CC BY: Attribution
- Роберт Беар и Дэвид Ринтул, Расширение законов о наследовании. 31 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m47304/latest/ . Лицензия : CC BY: Attribution
- Структурная биохимия/Хромосомы. Предоставлено : Wikibooks. Расположен по адресу : en.wikibooks.org/wiki/Structu…ry/Chromosomes . Лицензия : CC BY-SA: Attribution-ShareAlike
- аутосома. Предоставлено : Викисловарь. Расположен по адресу : en.wiktionary.org/wiki/autosome . Лицензия : CC BY-SA: Attribution-ShareAlike
- дикий тип. Предоставлено : Викисловарь. Расположен по адресу : en.wiktionary.org/wiki/wild_type . Лицензия : CC BY-SA: Attribution-ShareAlike
- Колледж OpenStax, Хромосомная теория и генетическая связь, 16 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44481/latest…e_13_01_01.jpg . Лицензия : CC BY: Attribution
- Роберт Беар и Дэвид Ринтул, Расширение законов о наследовании.31 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx. org/content/m47304/latest/ . Лицензия : CC BY: Attribution
- Колледж OpenStax, биология. 16 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44481/latest…ol11448/latest . Лицензия : CC BY: Attribution
- гомологичная рекомбинация. Предоставлено : Википедия. Расположен по адресу : en.Wikipedia.org/wiki/homolog…0recombination . Лицензия : CC BY-SA: Attribution-ShareAlike
- синапс. Предоставлено : Викисловарь. Расположен по адресу : en.wiktionary.org/wiki/synapsis . Лицензия : CC BY-SA: Attribution-ShareAlike
- связь. Предоставлено : Викисловарь. Расположен по адресу : en.wiktionary.org/wiki/linkage . Лицензия : CC BY-SA: Attribution-ShareAlike
- Колледж OpenStax, Хромосомная теория и генетическая связь, 16 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44481/latest…e_13_01_01.jpg . Лицензия : CC BY: Attribution
- Роберт Беар и Дэвид Ринтул, Расширение законов о наследовании.31 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m47304/latest/ . Лицензия : CC BY: Attribution
- Колледж OpenStax, Хромосомная теория и генетическая связь, 16 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44481/latest…e_13_01_02.jpg . Лицензия : CC BY: Attribution
- Колледж OpenStax, Хромосомная теория и генетическая связь, 16 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44481/latest…e_13_01_03.png . Лицензия : CC BY: Attribution
- Колледж OpenStax, биология. 16 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44483/latest…ol11448/latest . Лицензия : CC BY: Attribution
- кариотип. Предоставлено : Викисловарь. Расположен по адресу : en.wiktionary.org/wiki/karyotype . Лицензия : CC BY-SA: Attribution-ShareAlike
- транслокация. Предоставлено : Викисловарь. Расположен по адресу : en.wiktionary.org/wiki/translocation . Лицензия : CC BY-SA: Attribution-ShareAlike
- аутосома. Предоставлено : Викисловарь. Расположен по адресу : en.wiktionary.org/wiki/autosome . Лицензия : CC BY-SA: Attribution-ShareAlike
- Колледж OpenStax, Хромосомная теория и генетическая связь, 16 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44481/latest. ..e_13_01_01.jpg . Лицензия : CC BY: Attribution
- Роберт Беар и Дэвид Ринтул, Расширение законов о наследовании.31 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m47304/latest/ . Лицензия : CC BY: Attribution
- Колледж OpenStax, Хромосомная теория и генетическая связь, 16 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44481/latest…e_13_01_02.jpg . Лицензия : CC BY: Attribution
- Колледж OpenStax, Хромосомная теория и генетическая связь, 16 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx. org/content/m44481/latest…e_13_01_03.png . Лицензия : CC BY: Attribution
- NHGRI мужской кариотип человека. Предоставлено : Википедия. Расположен по адресу : en.Wikipedia.org/wiki/File:NH…_karyotype.png . Лицензия : Общественное достояние: Нет данных Авторские права
 16 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44481/latest…ol11448/latest . Лицензия : CC BY: Attribution
16 октября 2013 г. Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44481/latest…ol11448/latest . Лицензия : CC BY: Attribution  Расположен по адресу : en.wiktionary.org/wiki/autosome . Лицензия : CC BY-SA: Attribution-ShareAlike
Расположен по адресу : en.wiktionary.org/wiki/autosome . Лицензия : CC BY-SA: Attribution-ShareAlike  org/content/m47304/latest/ . Лицензия : CC BY: Attribution
org/content/m47304/latest/ . Лицензия : CC BY: Attribution 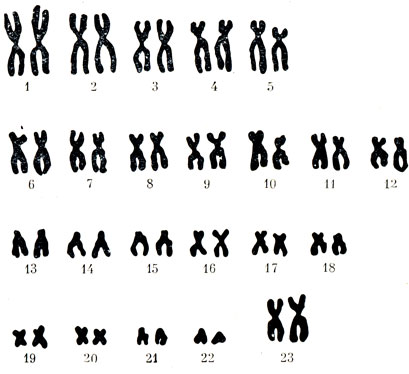 Предоставлено : Викисловарь. Расположен по адресу : en.wiktionary.org/wiki/linkage . Лицензия : CC BY-SA: Attribution-ShareAlike
Предоставлено : Викисловарь. Расположен по адресу : en.wiktionary.org/wiki/linkage . Лицензия : CC BY-SA: Attribution-ShareAlike  Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44481/latest…e_13_01_02.jpg . Лицензия : CC BY: Attribution
Предоставлено : OpenStax CNX. Расположен по адресу : http://cnx.org/content/m44481/latest…e_13_01_02.jpg . Лицензия : CC BY: Attribution  Расположен по адресу : en.wiktionary.org/wiki/karyotype . Лицензия : CC BY-SA: Attribution-ShareAlike
Расположен по адресу : en.wiktionary.org/wiki/karyotype . Лицензия : CC BY-SA: Attribution-ShareAlike 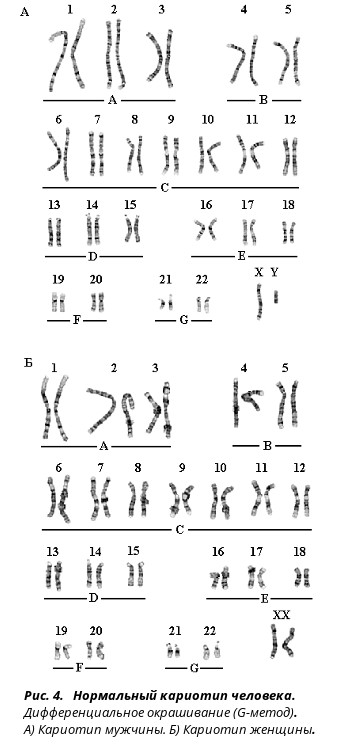 ..e_13_01_01.jpg . Лицензия : CC BY: Attribution
..e_13_01_01.jpg . Лицензия : CC BY: Attribution  org/content/m44481/latest…e_13_01_03.png . Лицензия : CC BY: Attribution
org/content/m44481/latest…e_13_01_03.png . Лицензия : CC BY: Attribution
Кариотипы человека для обучение: (46,XX, нормальная женщина) Эти кариотипы взяты у нормальной женщины.Eсть полный набор из 23 гомологичных пар. Эти кариотипы получены из лаборатории штата Висконсин.
гигиены. Они предназначены для использования в обучении, чтобы помочь
студенты изучают хромосомы человека. Каждый кариотип доступен в одной из трех форм:
Чтобы передать изображение на компьютер, нажмите кнопку подходящее имя изображения и сохраните его на жестком диске. диск. Для получения технической информации о методологии, с помощью которой эти мазки были созданы нажмите здесь. Кариотип_домУ вас есть предложения или исправления для этой страницы? Пожалуйста электронная почта Ларри Фелпса ([email protected]) © 1998 Попечительский совет университета Висконсинской системы. Нажмите здесь, чтобы отправить комментарии по электронной почте относительно BioWeb или его ссылок. |
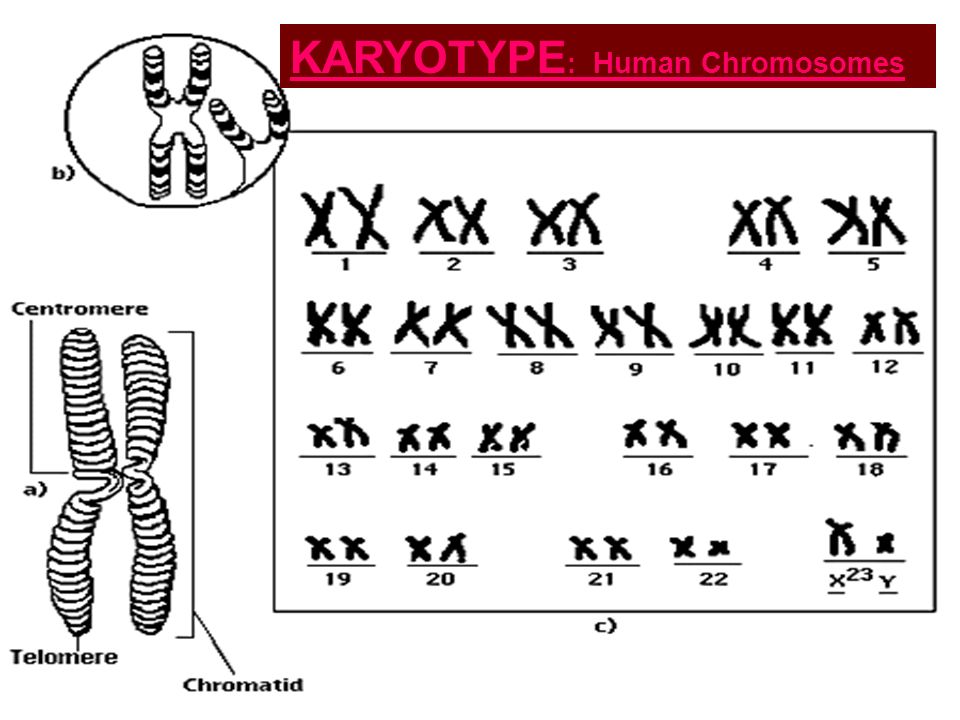 Авторские права на эти изображения
остается в Государственной лаборатории гигиены, обращайтесь к ним
для получения разрешения на любое другое использование, кроме некоммерческого
обучение.
Авторские права на эти изображения
остается в Государственной лаборатории гигиены, обращайтесь к ним
для получения разрешения на любое другое использование, кроме некоммерческого
обучение.
Диагностика выкидышей с помощью молекулярного кариотипирования: преимущества и недостатки
Было показано, что около 50% самопроизвольных абортов приходится на цитогенетические аномалии, причем наиболее частыми являются аутосомные трисомии и моносомии X. В этом исследовании общая частота аномалий составила 36% (35 из 96). Ни один из методов не смог идентифицировать все аберрации, при этом Т-образное окрашивание выявило 63% (22 из 35), а матричный CGH — 83% (29 из 35). Из 70 образцов, которые были успешно проанализированы обоими методами, в 55 (79%) обнаружены идентичные кариотипы (42 нормальных, 13 аномальных) и в 15 (21%) случаях выявлены расхождения.
В этом исследовании общая частота аномалий составила 36% (35 из 96). Ни один из методов не смог идентифицировать все аберрации, при этом Т-образное окрашивание выявило 63% (22 из 35), а матричный CGH — 83% (29 из 35). Из 70 образцов, которые были успешно проанализированы обоими методами, в 55 (79%) обнаружены идентичные кариотипы (42 нормальных, 13 аномальных) и в 15 (21%) случаях выявлены расхождения.
Опубликовано несколько исследований с использованием CGH или массива CGH для анализа самопроизвольных абортов.Этот широкий диапазон был вызван различиями в размерах исследований, отсутствием второго метода для обнаружения полиплоидии или использованием образцов, которые не смогли вырасти. В случае сравнения с традиционной цитогенетикой результаты обычно были согласующимися. Массив CGH в основном не смог обнаружить триплоидии, но выявил ряд небольших делеций и дупликаций и показал, что некоторые образцы 46, XX на самом деле были аномальными, но загрязнены материнскими клетками. Наше исследование имело те же характеристики, но имело высокий процент загрязнения материнскими клетками, что приводило к более низкому общему уровню аномалий (36%). Это загрязнение было причиной основного различия в результатах между обычным и молекулярным кариотипированием. Вероятно, есть две причины, по которым это загрязнение оказывает большее влияние на обычное кариотипирование: (1) Во время культивирования клеток может происходить отбор против хромосомных аномальных клеток27 (2) Матрица CGH лучше подходит для выявления слабовыраженного мозаицизма.27
Это загрязнение было причиной основного различия в результатах между обычным и молекулярным кариотипированием. Вероятно, есть две причины, по которым это загрязнение оказывает большее влияние на обычное кариотипирование: (1) Во время культивирования клеток может происходить отбор против хромосомных аномальных клеток27 (2) Матрица CGH лучше подходит для выявления слабовыраженного мозаицизма.27
Матрица CGH-анализ самопроизвольных выкидышей имеет преимущество по сравнению с традиционной цитогенетикой. Во-первых, не требуется культура клеток.Это позволяет избежать сбоя культивирования, контаминации культивирования, избыточного роста материнскими клетками или селекции против хромосомных аномальных клеток мозаичных плодов. Влияние чрезмерного роста материнских клеток можно увидеть в немного большем соотношении мужчин и женщин для массива CGH (17 из 100) по сравнению с Т-образным окрашиванием (10 из 100) среди случаев, идентифицированных как нормальные. Тем не менее, оба эти соотношения по-прежнему демонстрируют серьезное смещение в отношении соотношения полов. Эти результаты показывают, что образцы не только страдают от избыточного роста материнских клеток во время культивирования тканей, как видно из исследования Bell et al.,4 но образцы ворсинок также часто контаминированы материнскими клетками и что по-прежнему трудно получить точный образец фетального материала. Поэтому о результатах 46,XX всегда следует сообщать с осторожностью.
Эти результаты показывают, что образцы не только страдают от избыточного роста материнских клеток во время культивирования тканей, как видно из исследования Bell et al.,4 но образцы ворсинок также часто контаминированы материнскими клетками и что по-прежнему трудно получить точный образец фетального материала. Поэтому о результатах 46,XX всегда следует сообщать с осторожностью.
Дополнительным преимуществом исключения тканевых культур является сокращение времени выполнения цитогенетического анализа РОС. С перспективой дальнейшей автоматизации процесса CGH массива он станет только короче.
Предыдущие исследования показали способность массива CGH с массивами BAC обнаруживать слабый мозаицизм, установив более низкий порог около 10%.25,28,29 Мозаика самого низкого уровня описана при моносомии 7% 7.27 В этом исследовании мы выявили три случая с уровнем мозаицизма от 10% до 20%: мозаика образца 46, XY для изохромосомы 7p (рис. 1C), другой мужчина с 20% трисомией 13 и женщина с 10% трисомией 22 (рис. 1D). Кроме того, пять случаев были диагностированы как нормальные женские кариотипы с помощью традиционной цитогенетики, но позже были подтверждены FISH как моносомия X (Тернер) (рис. 1А) или нормальные мужские кариотипы (рис. 1В). На массиве эти два типа аномалий показали промежуточное значение log 2 для половых хромосом, но их можно было отличить друг от друга, особенно на Y-хромосоме.У пациентов Тернера отсутствовала Y-хромосома с соотношением log 2 от -2 к -4. Нормальные случаи XY, загрязненные материнскими клетками, показали лишь частично пониженные значения Y с отношением log 2 в диапазоне от -0,6 до -1,3. Используя этот метод, мы выявили случаи материнского заражения всего от 5 до 7% клеток 46,XY.
1D). Кроме того, пять случаев были диагностированы как нормальные женские кариотипы с помощью традиционной цитогенетики, но позже были подтверждены FISH как моносомия X (Тернер) (рис. 1А) или нормальные мужские кариотипы (рис. 1В). На массиве эти два типа аномалий показали промежуточное значение log 2 для половых хромосом, но их можно было отличить друг от друга, особенно на Y-хромосоме.У пациентов Тернера отсутствовала Y-хромосома с соотношением log 2 от -2 к -4. Нормальные случаи XY, загрязненные материнскими клетками, показали лишь частично пониженные значения Y с отношением log 2 в диапазоне от -0,6 до -1,3. Используя этот метод, мы выявили случаи материнского заражения всего от 5 до 7% клеток 46,XY.
Метод массива CGH позволил обнаружить все трисомии и моносомии, ранее идентифицированные с помощью Т-полосы. При использовании эталонной ДНК 47, XXY легче было наблюдать разницу между аномалиями половых хромосом, такими как трисомии и тетрасомии Х-хромосомы, и нормальными случаями 46, XX и 46, XY, что подтверждает предыдущие результаты Ballif et al.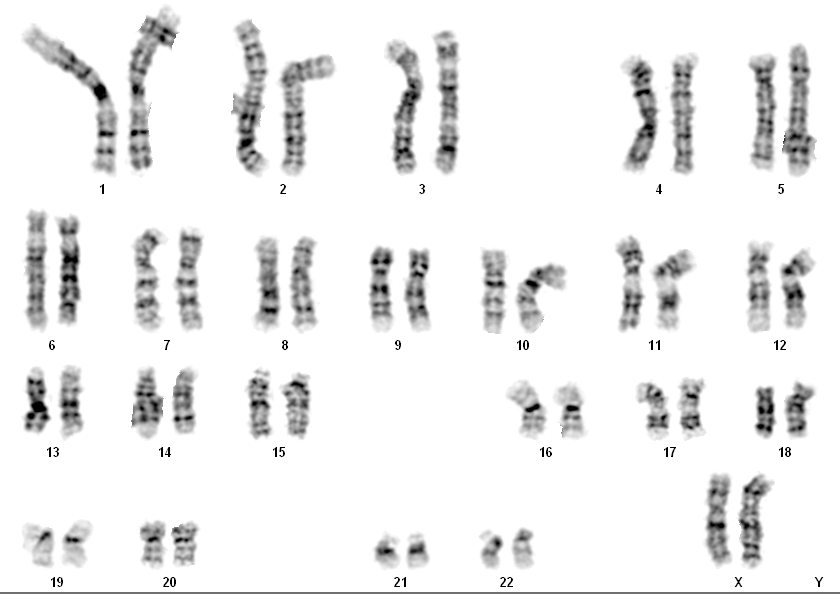 24 Также могут быть обнаружены некоторые триплоидии (XXY, а не XXX) (рис. 2, C и D) и некоторые тетраплоидии (все XXYY) (рис. 2E). Тетраплоидии создавали образец, аналогичный таковому у самцов, частично замаскированных материнским загрязнением (рис. 1В). Несмотря на наблюдения, сделанные в этих нескольких случаях, полиплоидия и сбалансированные транслокации обычно не обнаруживаются с помощью CGH на основе метафазы или матрицы. Другие группы справились с этим, выполнив анализы проточной цитометрии5 на полиплоидию. Теперь мы применяем интерфазную FISH с использованием зондов X и Y на некультивируемых тканях плода для выявления потенциальных триплоидий или тетраплоидий.Затем выполняется CGH массива. Эта комбинация позволяет идентифицировать все хромосомные дисбалансы, а также три- и тетраплоидии.
24 Также могут быть обнаружены некоторые триплоидии (XXY, а не XXX) (рис. 2, C и D) и некоторые тетраплоидии (все XXYY) (рис. 2E). Тетраплоидии создавали образец, аналогичный таковому у самцов, частично замаскированных материнским загрязнением (рис. 1В). Несмотря на наблюдения, сделанные в этих нескольких случаях, полиплоидия и сбалансированные транслокации обычно не обнаруживаются с помощью CGH на основе метафазы или матрицы. Другие группы справились с этим, выполнив анализы проточной цитометрии5 на полиплоидию. Теперь мы применяем интерфазную FISH с использованием зондов X и Y на некультивируемых тканях плода для выявления потенциальных триплоидий или тетраплоидий.Затем выполняется CGH массива. Эта комбинация позволяет идентифицировать все хромосомные дисбалансы, а также три- и тетраплоидии.
Массив профилей CGH с использованием ДНК нормального плода женского пола (A), нормального плода мужского пола (B), триплоидного плода XXX (C), триплоидного плода XXY (D) и тетраплоидного плода XXYY (E) по сравнению с ДНК из клеточной линии XXY. Для каждой панели ось x представляет клоны, упорядоченные от теломеры короткого плеча до теломеры длинного плеча, а хромосомы упорядочены от хромосом 1 до 22, X и Y.Ось y показывает log 2 трансформированных соотношений интенсивностей комбинированных экспериментов по замене красителя в каждом локусе (пациент Cy5/контрольный Cy3). Указаны хромосомы, которые не находятся в равных соотношениях.
Для каждой панели ось x представляет клоны, упорядоченные от теломеры короткого плеча до теломеры длинного плеча, а хромосомы упорядочены от хромосом 1 до 22, X и Y.Ось y показывает log 2 трансформированных соотношений интенсивностей комбинированных экспериментов по замене красителя в каждом локусе (пациент Cy5/контрольный Cy3). Указаны хромосомы, которые не находятся в равных соотношениях.
Массив CGH имеет более высокое разрешение, чем обычное кариотипирование, что позволяет обнаруживать небольшие делеции и дупликации, как это видно при обнаружении делеции стероидсульфатазы, описанной ранее. Разрешение недавно разработанных массивов неуклонно растет и ограничивается только размером клонов и расстоянием между клонами.Это дает возможность обнаруживать еще более мелкие делеции и дупликации, что позволит идентифицировать новые области или гены, играющие роль в раннем эмбриональном развитии. Однако массивы с повышенным разрешением обходятся дороже. В настоящее время стоимость выполнения низкоразрешающего массива CGH сопоставима со стоимостью кариотипирования.